 色谱分析(中国药科大学)第2章色谱法的基本参数及理论
色谱分析(中国药科大学)第2章色谱法的基本参数及理论

《色谱分析(中国药科大学)第2章色谱法的基本参数及理论》由会员分享,可在线阅读,更多相关《色谱分析(中国药科大学)第2章色谱法的基本参数及理论(19页珍藏版)》请在装配图网上搜索。
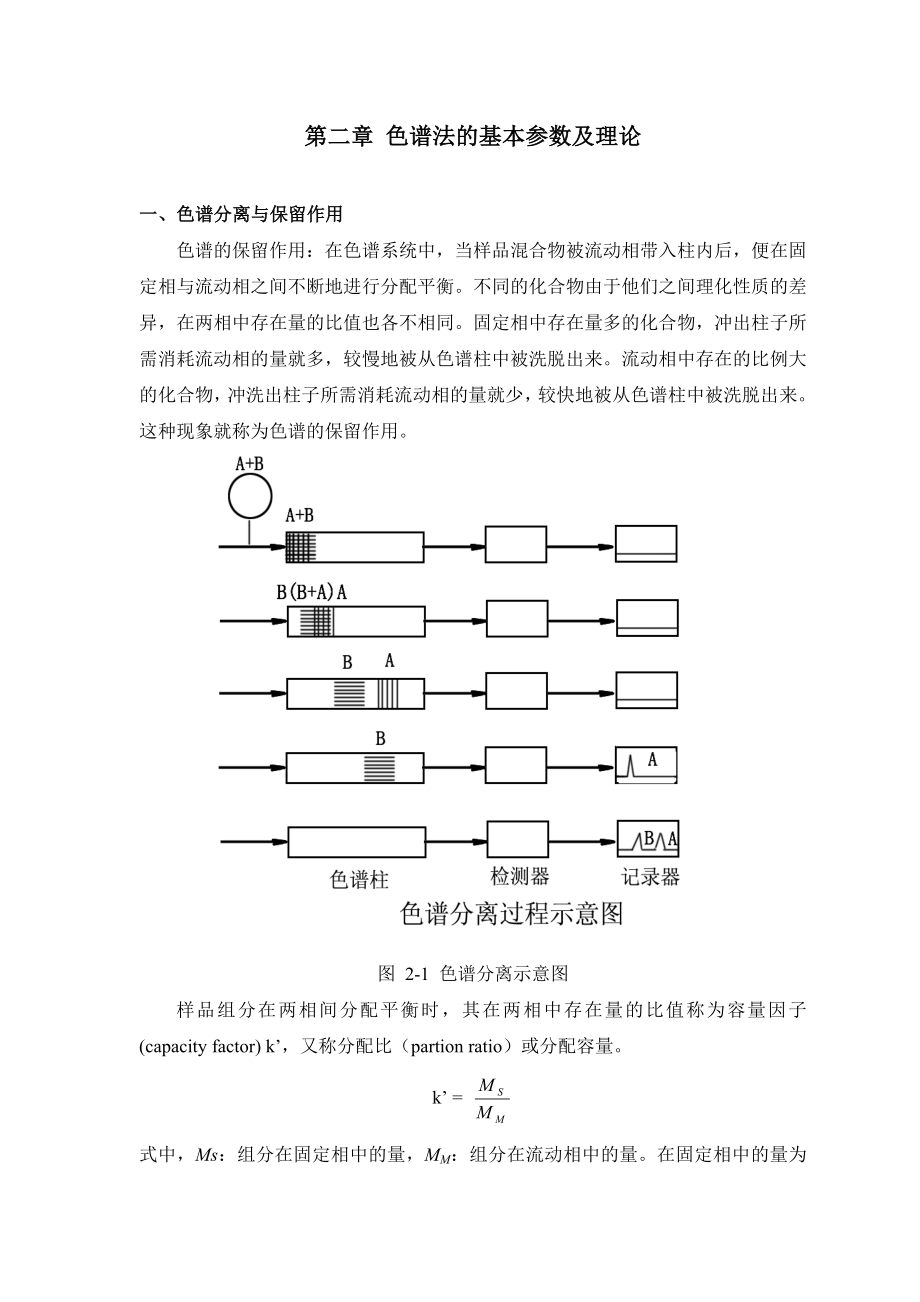
1、第二章 色谱法的基本参数及理论一、色谱分离与保留作用色谱的保留作用:在色谱系统中,当样品混合物被流动相带入柱内后,便在固定相与流动相之间不断地进行分配平衡。不同的化合物由于他们之间理化性质的差异,在两相中存在量的比值也各不相同。固定相中存在量多的化合物,冲出柱子所需消耗流动相的量就多,较慢地被从色谱柱中被洗脱出来。流动相中存在的比例大的化合物,冲洗出柱子所需消耗流动相的量就少,较快地被从色谱柱中被洗脱出来。这种现象就称为色谱的保留作用。图 2-1 色谱分离示意图样品组分在两相间分配平衡时,其在两相中存在量的比值称为容量因子(capacity factor) k,又称分配比(partion ra
2、tio)或分配容量。k = 式中,Ms:组分在固定相中的量,MM:组分在流动相中的量。在固定相中的量为零的化合物,其k=0,这些组分被称为在该色谱条件下的非保留物质。容量因子(分配比)可通过实验计算:k = 。即k为组分在固定相中消耗的时间与其在流动相中消耗的时间之比。样品组分在两相中分配平衡时,其在固定相和流动相中的浓度比称为分配系数(partion factor),分配系数以K表示。其公式如下:K = = k 式中,Ms/Vs为样品组分在固定相中的浓度,Mm/Vm为样品组分在流动相中的浓度。分配系数大的组分保留时间长(色谱的保留作用强),分配系数小的组分保留时间短(色谱的保留作用弱)。K
3、= k = k 式中 = 称为相比率,即色谱柱中流动相体积与固定相体积之比。例在毛细管GC中壁涂空心柱的相比为: = = = 式中r为毛细管柱横截面的半径,df为柱内壁固定液的膜厚。二、保留值1. 保留时间与保留体积保留时间(tR):从进样到柱后出现浓度最大值所需的时间。保留体积(VR):从进样到柱后出现浓度最大值所需消耗流动相的体积。VR = tRFc (Fc为流动相的流速)实验中Fc值的求法: HPLC直接从仪器读取 GC采用皂膜流量计在柱后测得 柱后载气肥皂液1020302. 死时间(tM)和死体积(VM)t M:不保留物质从进样开始到柱后出现浓度最大值所需时间。VM:不保留物质从进样开
4、始到柱后出现浓度最大值所需消耗流动相的体积。在以甲醇-水系统为流动相的反相HPLC中,甲醇峰的保留时间可近似看作死时间。在GC中,空气峰(GC-ECD中)的保留时间和甲烷峰(GC-FID中)的保留时间可近似看作死时间。(1) GC中tM的准确求算方法:tM = 式中tR(n+i)、tR(n)、tR(n-i)为同系物(如正构烷烃)中间隔碳原子数相等的组分的保留时间。Cn Cn-i = Cn+i Cn(2) HPLC中tM的测法:将流动相中溶剂峰峰的保留时间可近似看作死时间。3. 调整保留时间(tR)和调整保留体积(VR)tR为扣除死时间的保留值tR = tR - tM VR = VR VM k
5、= tR/ tM4. 相对保留值(relative retention volume)相对保留值1,2为被测组分的调整保留时间tR1与标准物的调整保留时间tR2之比。其公式如下:1,2 = = = = (k = tR/ tM)式中脚注号1表示被测物,2表示标准物。采用相对保留值,是为了消除某些操作条件,如流速和固定液流失等对保留值的影响。相对保留值中标准物可以是被测样品中的某一组分,也可以是人为地加入的某一化合物。1,2仅为柱温、固定液性质的函数,而与其他实验条件无关。1,2作为定性指标的缺点是标准物不统一,其数值随标准物的不同而不同。5. 保留指数(用于GC)保留指数(retention i
6、ndex)又称为柯瓦指数(Kovats index),是使用最广泛并被国际上公认的定性指标,它具有重现性好(精度可达0.1指数单位),标准物统一及温度系数小等优点。鉴于相对保留值存在标准物不统一,测量精度不够高等问题。Kovats提出了统一用正构烷烃系列作为标准物。把保留指数定义为:I x = 100 + 100N式中:I x为被测物的保留指数 tRx为被测物的调整保留时间 N为正构烷烃的碳原子数,如正己烷的N = 6 tRN及tR(N+1)分别为具有N个碳原子及(N+1)个碳原子的正构烷烃的调整保留时间。(1)保留指数的定义及公式来源保留指数:I(国际公认,用于GC中鉴定,定性),相对值,用
7、一系列正构烷烃为基准物。令正构烷烃的保留指数为I = 100N(N为正构烷烃的碳数)。被测物的保留指数Ix可用内插法算出。内插法的理论基础为:正构烷烃调整保留时间的对数(lg tR)与其相应的碳数成线性关系。l g tR = a N + b (1)设待测物x,其调整保留时间为tRx ,两正构烷烃n CN 及n CN+n的碳数分别为N和N+n,其调整保留时间分别为tRN及tR(N+n) 。tRN tRx tR(N+n)tRNtRXtR(N+n)设待测物x相当于正构烷的碳数为nx根据(1)式,则: lg tR(N+n) = a (N+n) + b (2) lg tRx = a nx + b (3)
8、 lg tRN = a N +b (4)(2) - (4) lg tR(N+n) - lg tRN = an (5)(3) - (4) lg tRx - lg tRN = a (nx N) (6)(6)/(5) = nx = N + n故待测物x的保留指数如下:Ix = 100 nx = 100 N + 100 n当相邻两个正构烷烃的碳数差值n = 1时,则 Ix = 100 N + 100(2)保留指数的特点 保留指数仅与固定相的性质及柱温有关,与其他实验条件无关,因此定性准确度比tR等保留值准确。习惯上将测定时的柱温写在I符号的下角,所用固定相写在I符号的上角。例如I,即采用SE-30为固
9、定相,柱温在150进行测试所得的保留指数。 同系物的保留指数与碳数呈线性关系。同系物中,相同的取代基有相同的保留指数增量。例:正构烷及正构醇的Ix Nc曲线如下:醇Ix=100Nc+b醇烷Ix=100Nc+b烷IxNcb烷=0两线平行b醇 b 烷 = 0,即相同碳数的正构醇总是比相同碳数的正构烷的Ix 高,且高出的值为一定值,即b醇。b醇为醇羟基(-OH)对保留指数做出的贡献。也即-OH使得醇类的色谱保留大于烷类(相同碳数时)。三、保留值与容量因子及分配系数的关系1. 保留值与容量因子k t R = tM(1 + k)由此可见容量因子k大的组分其保留时间长。例:在某色谱系统中,系统的死时间为0
10、.5,某样品组分在该系统中的容量因子为5,问该组分进样后需多长时间可出峰。解:tR = tM(1 + k) = 0.5(1+5) = 3 min2. 保留值与分配系数 tR = tM(1 + k) = tM(1 + KVS/ VM)可见,(1)K 大 tR 大。(2)在相同色谱条件下,如两组分的K值相同,则色谱峰重合;差别越大,色谱峰分离度越大。四、塔板理论(plate theory)为了研究色谱峰形状及评价柱效,建立了塔板理论。将色谱柱假想成由许多塔板所组成,在每个塔板上,组分在两相间达成一次平衡。 经多次分配平衡后,各组分由于分配系数不同而彼此分离。当塔板数足够多时,色谱流出曲线可用高斯(
11、Gaussian)分布表示:C = e 式中C为时间t时组分的浓度,W为被分离组分的重量,tR为对应于浓度极大点时的时间(即保留时间),为标准偏差。此方程的图形如下所示:(一)峰高及峰宽度峰高h为浓度极大值,即图中的AB。峰宽度可用以下三种方法表示:标准偏差:即图曲线拐点处宽度的一半,即EF的一半。半高峰宽Wh/2:(半峰宽,半宽度)即峰高一半处的宽度,如图中的GH,符号,Wh/2 = 2.354。峰宽Wb:从峰两边的拐点作切线与基线相交部分的宽度(I J),又称基线宽度。Wb = 4。(二)理论板数与板高柱效可用理论板数来表示,计算公式如下:n = 柱效通常用单位柱长的理论板数来表达,即每米
12、的理论板数(n/L)。柱效也可以用相当于一个理论塔板的高度表示,即:H = L/n该式的意义是一个理论板所占据的柱长,以毫米(mm)为单位。由于死时间的存在,n和H不能确切地反映柱效,尤其是对那些出峰早的组分,因此又提出了有效的塔板数及有效板高的概念。 = = N(Heff = 五、速率理论塔板理论对评价柱效是有用的,但没有讨论影响柱效的因素。Van Deemter等考察了多种影响柱效的因素,导出了如下关系式(Van Deemter曲线方程):H = A + B/ + C (1)式中,H为板高,为流动相的线速度。Van Deemter曲线方程描述了色谱峰柱内展宽的三种因素:涡流扩散项A;纵向扩
13、散项(或称分子扩散项)B/;传质速率项C 。1、 涡流扩散项A = 2 dp (2)式中, 为柱填充不规则因子;dp为填充物的平均颗粒直径。若柱子填装的不均匀或填料颗粒直径不均匀,流动相流过时就会由于流速不同而形成涡流扩散,使峰展宽。dp A ,n,峰展宽填充不均匀 A ,n,峰展宽2、 纵向扩散项(分子扩散项)试样分子在色谱柱内被流动相带向前时,由分子本身运动引起的轴向扩散也会引起峰展宽。 = (3)式中,Dg为样品组分在流动相中的扩散系数,为弯曲因子(亦称曲折性校正因子), 1。 Dg B/ H ,n , 峰展宽Dg与组分的性质,流动相的性质、温度、压力等有关。在GC中:增加载气密度,即增
14、加压力及载气分子量,可以降低Dg 。在HPLC中:分子在液体中的扩散系数比在气体中要小45个数量级,而流动相的线速度 10 mm/s。故在HPLC中B/很小,纵向扩散可忽略不计。 弯曲因子柱中填料的存在阻碍了分子扩散,故(3)式中引入曲折性校正因子 , 1,空心毛细管柱 = 1 。 流动相流速 B/ H ,n , 峰展宽。3、传质速率项C (又称传质阻力项)试样分子被流动相带着经过色谱柱时,分子不断的由流动相进入固定相,同时又从固定相不断进入到流动相。在这一过程中,由于传质速率不是很快,而流动相却有相当的流速,因此在两相的界面难以达到真正的快速平衡,这就造成了流动相中试样的分子较固定相中的试样
15、分子略微跑的快一点,这就引起了峰展宽。 实际浓度平衡浓度界面流动相固定相流动相流动方向C = + 流动相中的传质阻力 固定相中的传质阻力和分别为常数;dp为固定相的粒径;df为固定相的膜厚;Dm为组分在流动相中的扩散系数;Ds为组分在固定相中的扩散系数。有关传质速率项C : 在GC中:C = + 0.01式中DL即DS,Dg即Dm = 0.01 = 在HPLC中:C = + + :固定相微孔内的流动相,称为滞留区流动相,它是不流动的,固定相的微孔越小越深,传质速率就越慢,峰展宽也就越大。式中, + = , = Golay方程:对于毛细管柱,方程H = A + B/ + C 中,A 项不存在。G
16、olay方程:H = B/ + C ,毛细管柱 = 1,故B = 2Dg(1) df C ,H ,n ,峰展宽。70年代80年代,GC中固定液的涂布量一般为10 20% 减小df 现在GC中固定液的涂布量多用1 5%(2) Ds,Dm H , n (3) C 将H对作图,可得Van Deemter曲线:当为最佳流动相流速opt时,C opt = ,即 opt = ,但实际工作中为了节省时间,故采用的实际 opt 。Van Deemter 方程告诉我们:(1)要提高柱效,可 减小填料粒径; 提高填装均匀性; 减小固定液膜厚。(2)改变流速可获得最佳柱效。六、引起峰展宽的柱外因素引起峰展宽的柱外因
17、素主要为死体积。因此,降低柱外效应对板高的影响,可以从如下几方面着手:1. 尽量减小连接管线的长度。2. 采用细内径的管线为连接线。3. 采用小死体积的检测器。4. 进样技术、方式也会引起峰展宽。5. 进样体积也是引起峰展宽的柱外因素之一,体积 峰展宽。七、分离度及其影响因素1、分离度(Resolution)分离度(R)用以衡量相邻峰的分离情况,由下式计算R = = (注:W = 4,W1/2 = 2.354)式中tR1及tR2分别为组分1、2的保留时间,W1及W2分别为组分1、2的峰宽。W1/2(1),W1/2(2)分别为组分1、2的半峰宽。12W1W212tR1tR2对于两个等面积峰,R
18、= 1.5,称基线分离,两组分达99.7%的分离。又称6分离) R = 1,称基本分离,两组分达98%的分离。(又称4分离) R = 1.25,达99.2%的分离。实际工作中,中华人民共和国药典要求“除另有规定,分离度应大于1.5”。3、 影响分离度的因素由上式可知,分离度不仅取决于两组分的保留时间,也取决于其对应色谱峰的宽度。相同的分离度在高效或低效色谱系统中均可达到,因为,R与以下参数有关:R = k对R的贡献:R k增加,R增加 当k= 5 6时已足够,对分离度的贡献已基本上达到最大,再增加对分离度已无多大贡献。上式称“分离度方程”,式中n为理论塔板数,表示柱效;称分离因子(表示分离选择
19、性), = = k1, k2分别为两组分的容量因子。分离因子():代表两组分在相同色谱条件下的分离选择性。在GC中:(1)填充柱分离 低柱效(n)高分离因子()(2)毛细管分离高柱效(n)低分离因子() GC中,对于填充柱,其柱效低,因而为了达到预期的分离度,选择性是最重要的因素,因此,在填充柱GC色谱中,需提供许多种性质不同的固定相供选择。而在高效毛细管GC中,只需使用几种固定相就可分离复杂的混合物。4、 改善分离的一般原则:(1) 增加tR(= tR2 - tR1)(a) 增加柱长(b) 增加固定相的量(c) 选择分离因子较大的色谱条件 . 降低柱温 . 选择不同的固定相(填充柱GC中常用
20、) . 选择不同的流动相(HPLC中常用)改善分离(a)、(b)、(c)三种措施的理论基础如下:(a)增加柱长由公式R = 可以推导出对于长度不同的同一种色谱柱: = = (L1、L2分别为两根柱的长度)R2 = R1 可见,增加柱长L2,( ),可提高分离度(R2)。(b)增加固定相的量增加固定相的量,则k2 R (c)降低柱温对分离因子而言: = (Tb2 - Tb1)+ c式中,Tc为柱温,Tb1和Tb2分别为组分1、2的沸点,c为常数,R为热力学常数。可见,Tc R 。(2)减小谱带宽度(峰宽)W 提高柱效,减小塔板高度(a) 使用颗粒更细的填料(b) 更小心、均匀的填充色谱柱 减小色
21、谱系统的死体积如在填充经典的制备柱(植化中用)时,经常采用湿法装柱。柱子装好后一般要在流动相流动的情况下给出足够的时间(24 48 h),让固定相柱面不再沉降后,才能上样品。 减小进样量八、系统适应性试验(System suitability test)用规定的对照品对仪器进行试验和调整,应达到规定的要求;或规定分析状态下色谱柱的最小理论塔板数、分离度、重复性和拖尾因子。1、 色谱柱的理论塔板数(n)在选定的条件下,用对照品或内标计算色谱柱的理论塔板数,n = 5.54(2,若达不到规定的理论塔板数,则需更换色谱柱。2、 分离度定量分析时,为便于准确测量,要求定量峰与其他峰或内标峰之间有较好的
22、分离度。计算公式 R = ,除另有规定,R应大于1.5。3、 拖尾因子(tailing factor)为保证测量精度,特别当采用峰高法测量时,应检查待测物峰的拖尾因子(T)是否符合该品种项下的规定,或不同浓度进样的校正因子误差是否符合要求。计算公式:T = 式中,W0.05 为0.05峰高处的峰宽,d1为峰高极大至峰前沿之间的距离。除另有规定,T应在0.95 1.05之间。对称峰T = 1,拖尾峰T 1,前沿峰T 1。4、 重复性取各品种项下的对照品溶液,连续进样5次,除另有规定外,其峰面积测量值的相对标准偏差应不大于2.0%。也可按各品种校正因子测定项下,配制80%,100%和120%的对照
23、品溶液,加入规定量的内标溶液,配成3种不同浓度的溶液,分别进样3次,计算平均校正因子,其相对标准偏差不应大于2.0%。九、测定法定量测定时,可根据具体情况采用峰面积法或峰高法。测定杂质含量时,须采用峰面积法。1、内标法加校正因子测定含量按各品种项下规定,配成含内标的校正因子测定用对照溶液,进样分析,计算校正因子:校正因子f = 式中:AS为内标的峰面积或峰高,AR为对照品的峰面积或峰高。CS为内标的浓度,CR为对照品的浓度。再取各品种项下,含有内标的供试品溶液进样分析。计算含量:含量(Cx)= f式中,AX为供试品(或杂质)峰面积或峰高;Cx为供试品(或杂质)的浓度。f、AS、CS的意义同上。
24、2、外标法测定含量按各品种项下配制对照品溶液和供试品溶液,进样分析,计算含量:Cx = CR, 式中符号意义同上。3、面积归一化法该方法是测量各物质峰的面积和色谱图上除溶剂色谱峰以外的总色谱峰面积,计算各峰面积占总峰面积的百分率即得。该方法误差大(因为各物质在检测器上的响应不一定相同),只能粗略的考察供试品中各物质的含量,一般不用于微量杂质的检查。4、标准曲线法采用5个以上浓度点制备随行标准曲线,将待测样品的响应值代入标准曲线,计算含量。该方法主要用于各样品中待测物浓度相差很大时采用,如生物样品中待测物的高浓度样品与低浓度样品可能相差几十倍几百倍。标准曲线不一定过零点。5、 加校正因子的主成分
25、自身对照法该法在测定杂质含量时用。首先测定各杂质校正因子,再同“不加校正因子的主成分自身对照法”一样开展试验。计算杂质含量时,各峰面积均应乘以各自的校正因子后再计算。6、 不加校正因子的主成分自身对照法该法用于估算药物中的杂质的含量。将供试品溶液稀释成与杂质限度(常为1%)相当的溶液作为对照溶液,进样,调节仪器灵敏度,使对照品液主成分约达满量程的10% 25%。再分别进样供试品溶液及对照品溶液。测定供试品溶液色谱图上各杂质峰面积,并与对照液主成分峰面积比较,计算杂质含量。十、色谱等温线色谱等温线是分配系数K的图示方法,它反映了在柱温不变的情况下,样品组分的分配系数K随进样量的增加而变化的情况。
26、分配系数的表达式如下:K = ,分别为样品组分在固定相中的量和流动相中的量。以为横坐标,为纵坐标,即得色谱等温线。在气相色谱中通常有三种类型等温线表示色谱状态。CsCsCsCmCmCm线性等温线凸形等温线凹形等温线不变变小变大tR不随进样量而变tR随进样量增加而减少tR随进样量增加而变大正则峰拖尾峰伸舌峰(1)线性等温线:整个工作浓度范围K都保持不变,为一常数,谱带移动速度与浓度无关,故色谱峰的前沿与后沿是对称的,呈高斯分布。(2)凸形等温线:K值随浓度变化,浓度增大时,K值变小,溶质移动速度加快,因此,色谱峰有一个变锐的前沿和扩散的后沿。保留值随浓度的增加而减小。(3)凹形等温线:K值随浓度增大而变大,色谱峰有一个扩散的前沿和变锐的后沿,保留值随浓度的增加而增大。对称峰的保留值与进样量无关,非对称性峰的保留值与进样量有关。保留值随进样量变化的主要原因是溶质在气、液两相间分配的非线性。拖尾峰的保留值随进样量增加而减小,伸舌峰的保留值随进样量增加而增加。一般,把进样量外延至零时的保留时间作为标准值,以消除进样量多少对保留值的影响。
- 温馨提示:
1: 本站所有资源如无特殊说明,都需要本地电脑安装OFFICE2007和PDF阅读器。图纸软件为CAD,CAXA,PROE,UG,SolidWorks等.压缩文件请下载最新的WinRAR软件解压。
2: 本站的文档不包含任何第三方提供的附件图纸等,如果需要附件,请联系上传者。文件的所有权益归上传用户所有。
3.本站RAR压缩包中若带图纸,网页内容里面会有图纸预览,若没有图纸预览就没有图纸。
4. 未经权益所有人同意不得将文件中的内容挪作商业或盈利用途。
5. 装配图网仅提供信息存储空间,仅对用户上传内容的表现方式做保护处理,对用户上传分享的文档内容本身不做任何修改或编辑,并不能对任何下载内容负责。
6. 下载文件中如有侵权或不适当内容,请与我们联系,我们立即纠正。
7. 本站不保证下载资源的准确性、安全性和完整性, 同时也不承担用户因使用这些下载资源对自己和他人造成任何形式的伤害或损失。
