 现代电化学-第3章电极溶液界面的结构与性质.ppt
现代电化学-第3章电极溶液界面的结构与性质.ppt

《现代电化学-第3章电极溶液界面的结构与性质.ppt》由会员分享,可在线阅读,更多相关《现代电化学-第3章电极溶液界面的结构与性质.ppt(108页珍藏版)》请在装配图网上搜索。
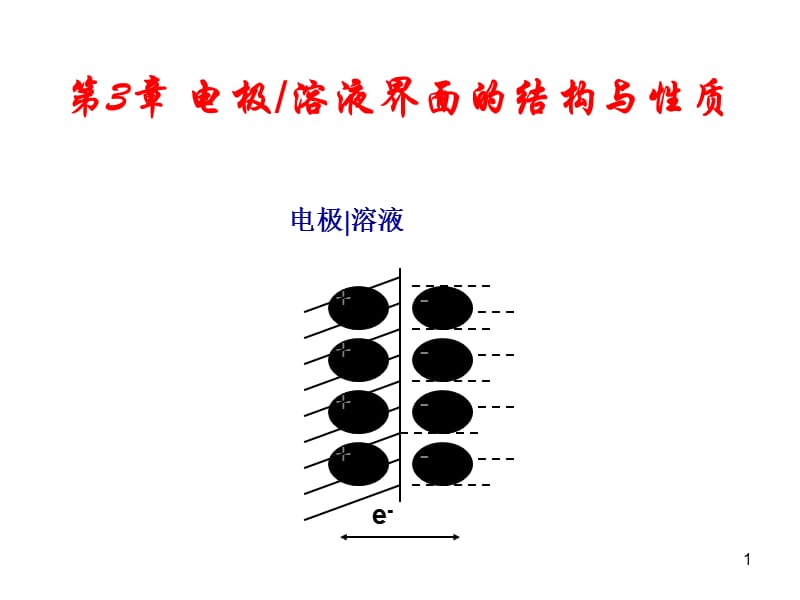
1、1,第3章 电极/溶液界面的结构与性质,2,3.1 研究电极溶液界面性质的意义,各类电极反应都发生在电极溶液的界面上,界面的结构和性质对电极反应有很大影响。,1界面电场对电极反应速度的影响,双电层电位差(即电极电位)为1 V,界面两个电荷层的间距为10-8cm时,其场强可达l08 Vcm,2. 电解液性质和电极材料及其表面状态的影响,析氢反应2H+ + 2e- H2在Pt电极上进行的速度比在Hg电极上进行的速度大107倍以上,3,3.2 电极溶液界面性质的研究方法,电极溶液界面: 两相之间的一个界面层,即与任何一相基体性质不同的相间过渡区域。,界面结构在这一过渡区域中剩余电荷和电势的分 布以及
2、它们与电极电位的关系。 界面性质界面层的物理化学特性,特别是电性质。,4,由于界面结构与界面性质之间有着密切的内在联系,因而研究界面结构的基本方法是测定某些重要的、反映界面性质的参数 (例如界面张力、微分电容、电极表面剩余电荷密度等)及其与电极电位的函数关系。把这些实验测定结果与根据理论模型推算出来的数值相比较,如果理论值与实验结果比较一致,那么该结构模型就有一定的正确性。,研究方法:,5,理想极化电极:在金属-溶液界面间不发生电荷转移(Hg电极),适合于进行界面研究的电极,给这个电化学系统施加一个电势,由于理想极化电极和溶液的界面间应该没有电子的转移,界面上不发生氧化还原等电极反应, 那么外
3、电源输入的全部电流都用于建立或改变界面结构和电极电势,这样,就可以方便地把电极电势改变到所需要的数值,并可定量分析建立这种双电层结构所需要的电量。,6,3.3 电毛细现象,3.3.1 电毛细曲线及其测定,对电极体系,界面张力()不仅与界面层的物质组成有关,而且与电极电势()有关。这种界面张力随电极电势变化的现象叫做电毛细现象。界面张力与电极电势的关系曲线叫做电毛细曲线。,通常用毛细管静电计测取液态金属电极的电毛细曲线,7,p 附加压力 表面张力(J/cm2) r 弯液面曲率半径 rc 毛细管半径 接触角 h 汞柱高度,8,电毛细曲线,汞溶液界面存在着双电层,即界面的同一侧带有相同符号的剩余电荷
4、。无论是带正电荷还是带负电荷,由于同性电荷之间的排斥作用,都力图使界面扩大,而界面张力力图使界面缩小,二者作用恰好相反。因此,带电界面的界面张力比不带电时要小,并且表面电荷密度越大,界面张力就越小。最高点处是电极表面剩余电荷密度为零时,其它点处表面带过剩电荷。,呈抛物线状, why?,9,3.3.2 电毛细曲线的微分方程,一般体系,Gibbs-Duham方程:,其中:ni 组分 i 的摩尔数, i 组分 i 的化学势。,存在相界面的体系,Gibbs-Duham方程:,其中:A 界面面积, 表面张力,,10,在恒温、恒压下:, i的表面吸附量(mol/cm2), Gibbs吸附等温式,11,一般
5、情况下,不带电的固相中没有可以自由移动而在界面吸附的粒子,因而对固液界面,化学势一项只需要考虑液相中的吸附粒子。但对电极电位可变的电极体系来说,可以把电子看成是一种能自由移动并在界面发生吸附的粒子。若电极表面剩余电荷密度为q(C/cm2),则电子的表面吸附量为:,其化学位变化为:,因此:,12,把电子这一组分单独列出来,则有:,理想极化电极的界面上没有化学反应发生,溶液中物质组成不变,即对于溶液中每一组分来说:di =0。,电毛细曲线的微分方程Lippman公式,Gibbs吸附等温式,13,表面电荷密度q=0时的电极电势,也就是与界面张力最大值相对应的电极电势称为零电荷电势,常用符号0表示。,
6、根据Lippman公式,可以直接通过电毛细曲线的斜率求出 某一电极电位下的电极表面剩余电荷密度q,做图就得到 q-曲线(II)。,零电荷电势0,14,(1)当电极表面存在正的剩余电荷时,根据Lippman公式,可以判断表面剩余电荷密度的符号,随电极电势变正,界面张力不断减小带正电。,(2)当电极表面存在负的剩余电荷时,随电极电位变负,界面张力也不断减小带负电,15,不论电极表面存在正剩余电荷还是负剩余电荷,界面张力都将随剩余电荷数量的增加而降低。,16,3.3.2 离子表面剩余量,溶液一侧剩余电荷密度:,式中:zi-离子i的价数 i溶液一侧离子i的表面剩余量,若保持和除i组分外的其他各组分j不
7、变,一定电极电位时的i离子表面剩余量(mol/cm2),17,当溶液成分发生变化时,由于参比电极电势变化而引起的电极电势变化为:,(电中性),(化学势加和性),其中相对为相对于浸在组成经历了同样变化的溶液中的参比电极测得的研究电极电势的变化。,若所用参比电极对于溶液中的正离子是可逆的(如氢电圾),则:,若参比电极对于溶液中的负离子是可逆的(如甘汞电极),则:,(等电量),18,若参比电极对负离子可逆,则:,若参比电极对正离子可逆,则:,参比电极对负离子可逆:,参比电极对正离子可逆:,相对,相对,19,具体求解离子表面剩余量的步骤: (1)测量不同浓度电解质溶液的电毛细曲线 (2)从各条电毛细曲
8、线上取同一相对电势下的值,作出-lna关系曲线 (3)根据-lna关系曲线,求出某一浓度下的斜率,zF,在0.1M各种电解质溶液中,20,3.4 双电层微分电容,电极/溶液界面的双电层可以当作平行板电容器来处理, 但必须注意的是界面双电层的电容并不完全像平行板电容器那样是恒定值,而是随着电极电势的变化而变化的。因此,应该用微 分形式来定义界面双电层的电容称为微分电容Cd。,从电毛细曲线求可求得微分电容值(F/cm2),21,Cd可以通过实验来测量,将测出不同下电极电势下的Cd值相对于绘图, 得到的曲线称为微分电容曲线。,滴汞电极在不同浓度KCl溶液中的微分电容曲线,滴汞电极在不同无机盐溶液中的
9、微分电容曲线,22,已知:q = 0时, = 0, 以此为边界条件进行积分, 可得:,电极电势为时的q值相当于图中的阴影部分。,q,求q 值时,微分电容法比毛细曲线法相比更为精确和灵敏。电毛细曲线的直接测量只能在液态金属电极上进行,而微分电容法测量还可以在固体电极上直接进行。,23,积分电容:从 0到某一电位之间的平均电容称为积分电容 。 与 的关系: ,24,3.5 双电层的结构,在电极溶液界面存在着两种相间相互作用: (1)电极与溶液两相中的剩余电荷所引起的静电长程作用;(2)电极和溶液中各种粒子(离子、溶质分子、溶剂分子等等)之间的短程作用,如特性吸附、偶极子定向排列等,它只在几个的距离
10、内发生。 这些相互作用决定着界面的结构和性质,双电层结构模型的提出都是从这些相互作用出发的。在距电极表面距离不超过几个的“内层”中,需要同时考虑上述两种相互作用;而在距电极表面更远一些的液相里的“分散层”中,只需要考虑静电相互作用。,25,电极溶液界面的基本结构,静电作用使得符号相反的剩余电荷力图相互靠近,形成紧密的双电层结构,简称紧密层。 热运动处使荷电粒子倾向于均匀分布,从而使剩余电荷不可能完全紧贴着电极表面分布,而具有一定的分散性,形成分散层。,电极溶液界面的紧密双电层结构,考虑了热运动干扰时的电极溶液界面双电层结构,26,剩余电荷分布的分散性决定于静电作用与热运动的对立统一结果,因而在
11、不同条件下的电极体系中,双电层的分散性不同。 在金属相中,自由电子的浓度很大,可达1025 mol/dm3 ,少量剩余电荷在界面的集中并不会明显破坏自由电子的均匀分布,因此可以认为金属中全部剩余电荷都是紧密分布的,金属内部各点的电位均相等。,在溶液相中,当溶液总浓度较高,电极表面电荷密度较大时,由于离子热运动较困难,对剩余电荷分布的影响较小,而电极与溶液间的静电作用较强,对剩余电荷的分布起主导作用,溶液中的剩余电荷也倾向于紧密分布,形成紧密双电层。,27,如果溶液总浓度较低,或电极表面电荷密度较小,那么离子热运动的作用增强,而静电作用减弱,形成紧密与分散层共存的结构。 如果由半导体材料和电解质
12、溶液组成电极体系,那么在固相中,由于载流子浓度较小(约为1017 mol/dm3 ),则剩余电荷的分布也将具有一定的分散性。,金属与稀溶液界面的双电层结构,半导体与稀溶液界面的双电层结构,28,双电层由紧密层和分散层组成。 图中d为紧贴电极表面排列的水化离子的电荷中心与电极表面的距离,也就是离子电荷能接近电极表面的最小距离。 从x=0到x=d的范围内,不存在剩余电荷,即为紧密层。 若假定紧密层内的介电常数为恒定值,则该层内的电位分布是线性变化的。 从x=d到剩余电荷为零(溶液中)的双电层部分即为分散层。其电位分布是非线性变化的。,电极溶液界面上电势及剩余电荷的分布情况,29,1电位:离子电荷能
13、接近电极表面的最小距离处的平均电位。 在没有考虑紧密层内具体结构的情况下常习惯于把1电位定义为距离电极表面一个水化离子半径处的平均电位。 在不同结构的紧密层中,d的大小是不同的,所以把1电位看成是距离电极表面d处,即离子电荷能接近电极表面的最小距离处的平均电位更合适些。 也可把1电位看作紧密层与分散层交界面的平均电位。,30,若以a表示整个双电层的电位差,则紧密层的电位差为(a - 1),分散层的电位差1。 a 和 1均是相对于溶液深处的电位(规定为零)而言的。 双电层电容看作串连模型。可用下式计算双电层电容:,31,3.5.1 双电层结构模型,1. Helmholtz模型(1879),“平板
14、电容器”模型或“紧密双电层”模型。,电极表面上和溶液中的剩余电荷都紧密地排列在界面两侧,形成类似平板电容器的界面双电层结构(金属电极/高浓度溶液时)。,紧密层,该模型基本上可以解释界面张力随电极电势变化的规律和微分电容曲线上零电荷电势两侧各出现一个平台区,但是,无法解释为什么在稀溶液中微分电容曲线上零电荷电势处会出现极小值,也没有触及微分电容曲线的精细结构(即电容随电极电势和溶液浓度变化而变化)。,-电荷密度 -介电常数,32,2. Gouy-Chapman模型(扩散层模型)(1910-1913),扩散层,零电荷电势,无紧密层,溶液中的离子在静电作用和热运动作用下,按势能场中粒子的分配规律(B
15、oltzmann分布律)分布在邻近界面的液层中,即形成“分散层”。分散层中的电势与距离呈曲线关系。 按照这种模型,并假设离子电荷为理想的点电荷,可以较满意地解释稀溶液中零电荷电势附近出现的电容极小值。但这种模型完全忽略了紧密层的存在,因而当溶液浓度较高或表面电荷密度值较大时,计算得出的电容值远大于实验测得的数值,而且解释不了微分电容曲线上“平台区”的出现。,33,3. Gouy-Chapman模型方程,-相对于本体溶液的电势 e-电子的电荷 k-波尔兹曼常数 T-绝对温度 ni0本体粒子数浓度 zi离子价数(电荷数),离子分布服从Boltzmann定律,把溶液分别成平行于电极的若干薄层,其厚度
16、为dx,所有这些薄层彼此处于 热平衡。因为静电电势在的变化,任意物质i的离子在各个薄层并不具有相同的能 量。薄层可以认为是等量衰减的能态;则任意薄层中离子的总数为:,(1),34,(2),假设离子电荷是连续分布的,从静电学的Poisson方程可知:,任意薄层中单位体积的总电荷(剩余电荷的体积密度C/m3) :,(3),35,将(2)带入(3)整理可得Poisson-Boltzmann 方程:,(4),利用数学关系式:,(5),比较(4)和(5)可得:,(6),36,对(6)积分可得:,远离电极表面,x = 时: = 0,(d/dx)=0,可求得常数,于是有:,(7),(8),对于z:z型电解质
17、,(8)式简化为:,(9),37, 分散层中电势分布:,将(9)式整理并积分, x=0, =0 ,则有:,(10),(11),或:,式中:,(12),(13),穿越整个扩散层的电势降,式(11)给出了扩散层电势与据电极表面的距离x之间的关系,也就是扩散层中电势分布情况。,38,电势总是从表面不断地逐渐衰减。在较大的0之下(剩余电荷密度大),分散层比较紧密(薄),电势下降很快。当0较小时, 衰减比较缓慢, 在0 小到某种极限时(25,0 50/z mV时), 0的衰减接近指数形式。,由模型方程可计算电势分布,39,对于稀水溶液体系, = 78.49 ,在T = 298K, ni0粒子数浓度用摩尔
18、浓度C代替,可得 :,C*- 电解质的本体浓度 (mol/L) 1/ -分散层特征厚度( cm),1:1,(13),表给出了1:1型 电解质的几种浓度下1/K值。显然电解质浓度越高时,扩散层越薄,随着电解液浓度的降低时,扩散层变的越来越较厚。从扩散层形成的原因来看,这显然是正确的。 另外,显然扩散层的厚度是非常小的,在通常的浓度下,0.1M,只有几个埃。,40, 剩余电荷密M和扩散层电势0的关系:,设想在所研究的体系中放置盒状的高斯表面,如图所示。盒子一端位于界面上。盒子的另一端延伸至场强d/dx基本上 为零的足够远的溶液中。这样,盒子中包括了分散层中所有的电荷, 根据高斯定律,其电量为:,(
19、14),为表面上各点的场强,除电极/溶液界面(x=0)处外,均为0,界面上(x=0)各点的场强为,41,(15),-溶液一侧电荷密度,(16),(9),将(9)代入(16)得:,42,对于稀水溶液体系,在T = 298K:,M剩余电荷密度(C/cm2) C*电解质的本体浓度 (mol/L),电极表面剩余电荷密度与扩散层电势差、溶液浓度、温度之间的关系。,(17),(18),式(17)即为电极表面剩余电荷密度与扩散层电势差和溶液浓度及温度之间的关系。利用这个公式,可以计算电极表面剩余电荷密度,或由剩余电荷密度计算扩散层电势差。,43, 微分电容:,将式(17)对扩散层电势降0求微分:,微分电容与
20、扩散层电势差、溶液浓度、温度之间的关系。,(19),对于稀水溶液体系,在T = 298K:,(20),Cd 微分电容(F/cm2) C*电解质的本体浓度 (mol/L),44,1:1电解质,298K,由(19)式计算的Cd曲线,微分电容曲线在零电荷电势附近出现的电容极小值,与实验结果吻合,但没有预测到微分电容曲线上“平台区”的出现,45,4. Stern模型(1924),Gouy-Chapman-Stern(GCS)模型,溶液中离子受到电极表面的库仑静电力和热运动双重作用,库仑力试图使离子整齐的排列在电极表面附近,而热运动则力图使其均匀的分布在溶液中,这两种作用互相抗衡的结果是:部分电荷在靠近
21、电极表面处形成紧密层,另一部分电荷分布在离电极表面稍远处形成扩散层。 Stern 模型较好的反映了界面双电层的真实结构,可以较满意地解释电容微分曲线上在零电荷电势附近出现的电容极小值和两侧出现“平台”的实验事实。,46,5. GCS模型方程,GCS模型, 电势分布方程,(21),由G-C模型推导出的关扩散层方程对于stern模型中的扩散层同样适用。,(10),47,积分整理:,(22),GCS模型中扩散层的电势分布,变化规律与G-C模型的扩散层是一致的。,根据Helmholtz模型,紧密层类似平行板电容器,电势分布是线性的。,Stern模型中电势分布先直线下降,然后曲线下降,48, 剩余电荷密
22、M和双电层电势0的关系:,据(9), 在x2处电场强度:,(23),(24),溶液一侧所有电荷都分布于扩散层中,由式(17)得:,(25),将式(24)代入式(25),则:,剩余电荷密M和整个双电层电势差0间的关系,(26),紧密层中线性分布,49, 微分电容,将式(26)微分并整理可得微分电容:,(27),(28),Cd由较小的电容所决定。在低浓度电解液体系,方程预见到在零电荷电势处Cd出现极小;在较高浓度的电解液中,CD变大到对Cd没有贡献, Cd由CH决定,为恒定值(平台)。,CH紧密层电容,CD扩散层电容。,50,Stern模型能比较好地反映界面结构的真实情况。但是,该模型在推导GCS
23、方程式时作了一些假设: (1) 把离子电荷看成点电荷并假定电荷是连续分布的; (2) 假设介质的介电常数不随电场强度变化; (3) 只简单地把紧密层描述成厚度不变的离子电荷层,忽略了紧密层组成的细节及由此引起的紧密层结构与性质上的特点。 因此,GCS双电层方程式对界面结构的描述只能是一种近似的、统计平均的结果,而不能用作准确的计算。,可以较满意地解释电容微分曲线上在零电荷电势附近出现的电容极小值和两侧出现“平台”的实验事实。,51,6. Grahame,Bockris, Devanathan, and Muller 模型,对stern 模型进行了补充和修正。这种补充和修正主要是考虑两个方面,一
24、个是溶剂化(水化)作用,一个是离子的吸附。,52,水分子是强极性分子,能在带电的电极表面定向吸附,形成一层定向排列的水分子偶极层。这个吸附在电极表面上的水分子偶极层覆盖度可达70以上,好像电极表面被水化了一样。 因而在通常情况下,紧靠电极表面的第一层是定向排列的水分子 偶极层,第二层才是由水化离子组成的剩余电荷层。偶极层的水分子由于在强界面电场中定向排列而导致介电饱和。,偶极层水分子的相对介电常数降低到56。 紧密层即离子周围的水化膜的相对介电常数上升到40左右。 通常水的相对介电常数为78。,53,离子与电极表面由于短程相互作用而发生物理或化学吸附,这种吸附与电极材料、离子本性及其水化程度有
25、关,被称为特性吸附。 溶液中的离子除了因静电作用而富集在电极/溶液界面外,还可能由于与电极表面的短程相互作用而发生物理吸附或化学吸附。这种吸附与电极材料、离子本性及其水化程度有关,被称为特性吸附。大多数无机阳离子不发生特性吸附,只有极少数水化能较小的阳离子,如Tl+,Cs+等离子能发生特性吸附。反之,除了 F-离子外,几乎所有的无机阴离子都或多或少地发生特性吸附。 有无特性吸附紧密层的结构是有差别的。,54,当电极表面带负电时,双电层溶液一侧的剩余电荷由阳离子组成。由于大多数阳离子与电极表面只有静电作用而无特性吸附作用,面且阳离子的水化程度较高, 所以,阳离子不容易脱出水化膜而突入水偶极层。这
26、种情况下的紧密层将由水偶极层与水化阳离子 层串联组成,称为外紧密层。 这些最近的溶剂化离子中心的位置称为外亥姆荷茨平面 (OHP), 它的厚度为从电极表面处到水化阳离子电荷中心的距离。若设x1 为第一层水分子层的厚度、 x2为 一个水化阳离子的半径,则 d x1+x2.,55,如果阴离子水化程度较低,阴离子就有可能够溢出水化膜,如果这个阴离子能进行特性吸附的话,那它就会取代水偶极层中的水分子而直接吸附在电极表面上,这些吸附离子与水偶极子等组成内紧密层。 阴离子电荷中心所在的液层称为内亥姆荷茨(IHP)平面。由于阴离子直接与金属表面接触,故内紧密层的厚度仅为一个离子半径比外紧密层厚度小很多。 根
27、据内紧密层与外紧密层厚度的差别解释微分电容曲线上为什么q0 时的紧密层 (平台区)电容比 q 0时大得多。,56,外紧密层的电容等效为水偶极层电容与水化阳离子层电容的串联。,外紧密层的电容只取决于水偶极层的性质,与阳离子种类无关,因而接近于常数。,57,外紧密层以外,就是由于静电力和热运动共同作用导致的扩散层(三维区域),从OHP到本体溶液。 有无特性吸附,紧密层的结构是有差别的,因此双电层的结构和性质也就不同,最直接的就是电势分布。 无特性吸附时,电势变化曲线象这个图。当有特性吸附时,虽然根据吸附的情况不同,电势变化也不同,但吸附对双电层的影响是确定的。,无特性吸附,58,3.5.2 零电荷
28、电位,零电荷电位 :电极表面剩余电荷为零时的电极电位 。由于电极表面不存在剩余电荷时,电极溶液界面就不存在离子双电层,所以也可将零电荷电位定义为电极溶液界面不存在离子双电层时的电位。 与 不同的原因:剩余电荷的存在是形成相间电位的重要原因,但不是的唯一原因。任何一相表面层中带电粒子或偶极子的非均匀分布也会引起相间电位。 零电荷电位仅仅表示电极表面剩余电荷为零时的电极电位,而不表示电极溶液的相间电位或绝对电位的零点。,59,曲线法测零电荷电位: 溶液越稀,微分电容最小值越明显。,铅、,60,的重要性,判断电极表面剩余电荷q的符号和数量 例:对于体系,61,在 处,一切依赖于q的表面性质均达极限值
29、 ,所以 是个特征点 。 双电层中的电位分布、界面电容、界面张力、各种粒子在界面的吸附行为、溶液对电极的润湿性和气泡在电极上的附着等。 将 选作新的电位衡量点,就有了一个新的衡量电极电位的体系零标电位。 零标电位:相对于零电荷电位的相对电极电位,以零电荷电位作为零点的电位标度。双电层电位差就是零标电位。 用零标电位研究电化学热力学是不适宜的。不同体系的零标电位不能通用,没有可比性。,62,室温下水溶液中的零标电位,63,64,3.5.3 电极/溶液界面的特性吸附,由于电极溶液界面上存在着一定范围内连续变化的电场,电极溶液界面的吸附现象比一般界面吸附更为复杂,有它自己特殊的规律性。,当电极表面带
30、有剩余电荷时,会在静电作用下使带相反符号电荷的离子聚集到界面区-静电吸附。 溶液中的各种粒子由于非静电作用力面发生吸附-特性吸附。,65,凡是能在电极溶液界面发生吸附面使界面张力降低的物质就叫表面活性物质(原子、分子、离子)。 表面活性粒子的吸附力(非静电作用力)-短程作用力,包括镜象力、色散力等物理作用和类似于化学键的化学作用。 特性吸附行为取决于电极与表面活性粒子之间、电极与溶剂分子之间、表面活性粒子与溶剂分子之间的相互作用。,不同的物质发生特性吸附的能力不 同、同一物质在不同的电极体系中的吸附行为也不相同。,66,电极溶液界面的吸附现象对电极过程动力学有重大的影响 (1) 表面活性粒子不
31、参与电极反应时,它们的吸附会改变电极表面状态和双电层中电位的分布,从而影响反应粒子在电极表面的浓度和电极反应的活化能,使电极反应速度发生变化。 (2)当表面活性粒子是反应物或反应产物(包括中间产物)时,就会直接影响到有关步骤的动力学规律。,利用界面吸附现象对电极过程的影响来控制电化学过程。,67,阴离子的吸附与电极电位有密切关系,吸附主要发生在比零电荷电位更正的电位范围,电极电位越正,阴离子的吸附星也越大。 在同一种溶液中,加入相同浓度的不同阴离子时,同一电位下界面张力下降的程度不同。 界面张力下降越多,表明该种离子的表面活性越强。在汞电极上的吸附能力(表面活性) : SO42-OH-Cl-B
32、r- I-S2- 。 阴离子吸附使电毛细曲线最高点,即零电荷电位向负方向移动,表面活性愈强的离子引起的负移的程度也愈大。,1. 无机离子的吸附 (1)阴离子吸附与电毛细曲线,68,电极表面没有剩余电荷,也没有剩余吸附时,电极溶液界面上不存在离子双电层,电极电位就是零电荷电位。 若发生阴离子特性吸附,吸附阴离子与溶液阳离子的静电作用形成吸附双电层( )。使得有吸附双电层的零电荷电位比没有特性吸附时向负移动 。 因为吸附双电层式建立在溶液一侧的,其电位差分布在分散层中,因而有1= 。,69,(2)吸附与电势分布 当电极表面带有正的剩余电荷时,因阴离子的特性吸附使得紧密层中负离子电荷超过了电极表面的
33、正剩余电荷-超载吸附。 超载吸附导致紧密层中过剩的负电荷又静电吸引溶液中的阳离子,形成三电层结构(正-负-正)。,70,无持性吸附(EEz)时,扩散层的电位的符号与总的电极电位符号相反。,71,(3)吸附与微分电容曲线,阴离子吸附时将脱去水化膜,挤进水偶极层,直接与电极表面接触形成内紧密层结构,从而使紧密层有效厚度减小,微分电容值增大。所以,在零电荷电位附近和比零电荷电位正的电位范围内。微分电容曲线比无特性吸附时升高。,72,“电极/溶液”界面模型概要(总结):,由于界面两侧存在剩余电荷(电子及离子电荷)所引起的界面双电层包括紧密层和分散层两部分。 紧密层是带有剩余电荷的两相之间的界面层,厚度
34、不超几个埃;而分散层是液相中具有剩余离子电荷及电势梯度的表面层,稀溶液中及表面电荷密度很小时厚度可达几百埃。 但在浓溶液中及表面电荷密度不太小时几乎可以忽视分散层的存在,即可近似认为分散层中的剩余电荷均集中在界面层的外表上。,73,分散层是由于离子电荷的热运动引起的,其结构(厚度、 电势分布等)只与温度、电解质浓度(包括价型)及分散层中的剩余电荷密度有关,而与离子的个别特性无关。 紧密层的性质决定于界面层的结构,特别是两相中剩余电荷能相互接近的程度。 大多数无机阳离子剩余电荷由于水化程度较高,且不能与电极表面上的金属原子发生化学相互作用,故不能逸出水化球而直接吸附在电极表面上,此时紧密层较厚。
35、 多数无机阴离子由于水化程度较低,特别是能与电极表面原子发生类是生成化学键的相互作用,它们往往能直接吸附在电极表面上而组成更薄的紧密层(此时为特性吸附)。,74,能在电极表面“特性吸附”的阴离子往往在电极表面上“超载吸附”。当出现“超载吸附”时,紧密层中的电势降与分散层中的电势降方向相反。此时界面结构及其中电势分布具有“三电层”形式。 由于分散层中的离子剩余电荷处在完全无序的热运动状态,可以统计地认为每一与电极平行的面上各点具有等电势。然而特性吸附的阴离子具有相对稳定的表面位置,因此特别是当这些阴离子的表面覆盖度不大时,所在平面上各点的电势具有二维的不均匀性。,75,OHP,IHP,OHP,d
36、1,d2,IHP,OHP,d1,d2, vs PZC,0,x,0 d1 d2,OHP,76,吸附的分类,静电吸附:静电作用下,异号离子相互吸引产生吸附。 特点:与电荷符号、数量直接相关 , 即: 非特性吸附:这类吸附是有机物的共性。 特点:与界面无关(即无选择性),只与吸附物质特性有关,即非物理吸附也非化学吸附。,77,特性吸附(选择性吸附):该类吸附由短程力所致。分为物理吸附和化学吸附两种。 物理吸附:短程力为镜像力、色散力、金属表面偶极层与极性分子作用、近程排斥力(范德华力)等; 化学吸附:本质是金属与吸附粒子间的不完全电子转移,形成吸附键,进而可形成表面化合物。 特点:有选择性,78,如
37、果形成双电层时只涉及荷电粒子之间的库伦引力,则影响界面电性质的因素不外乎是电解质的价型、浓度、溶剂化离子的大小以及相对于零点电位而确定的电极电位。 按照GCS理论,在同浓度、同价型的电解质溶液中测得的电毛细曲线和微分电容曲线应基本相同。若加入少量中性分子,只要不严重影响离子的活度系数及溶液的介电常数,也就不会影响界面的电性质。 除了表面剩余电荷引起的离子的静电吸附外,还有各种表面活性粒子的吸附现象。这比无机离子对电极过程的影响更为显著。,2. 有机物吸附对界面结构与性质的影响,79,2. 有机物吸附对界面结构与性质的影响,叔丁醇浓度: 10.00M 20.01M 30.05M 40.10M 5
38、0.20M 60.40M,绝大部分能溶于水的有机分子在界面上都具有程度不同的表面活性。 阴离子型:各种有机磺酸盐和硫酸盐;阳离子型:各种季铵盐;非离子型:环氧乙烷与高级醇的缩合物。,在电化学体系中用作添加剂,通过表面吸附控制电极过程,如各种缓蚀剂、光亮剂、表面润滑剂等。 随着叔丁醇浓度的增加,在电毛细曲线零电荷电势附近,界面张力下降,零电荷电势向正电势方向移动,表明吸附发生在零电荷电位附近的一定电位范围内。,1M NaCl溶液,80,在微分电容曲线零电荷电势附近,微分电容Cd值下降,随着有机物吸附覆盖度增大,微分电容降低得越多。两侧则出现电容峰值。,Cd随下降的原因:有机分子的介电常数比水小,
39、分子体积比水大,当有机分子取代水分子吸附在电极表面时,由Cd0/d 可知, Cd下降。,1 无有机物 2 达到饱和吸附 3 未达饱和吸附,随着有机分子浓度的增加,吸附覆盖度也将增 大,因而微分电容减少得更多。当电极表面全部被有机分子吸附 覆盖(饱和吸附覆盖=1)时所测得的电容值全部是被覆盖表面的电容,因而出现极限值。,81,假设电极表面上被覆盖的部分与未覆盖的部分是彼此互不影响的:,(1),q电极表面部分被覆盖时的电荷密度 q=1完全覆盖的表面上的电荷密度 q=0未覆盖时的表面上的电荷密度,设覆盖表面和未覆盖表面上紧密双电层中的电势相同, 对式(1)微分可得:,(2),C=1和C=0完全覆盖表
40、面和未覆盖表面的界面微分电容,82,在微分电容曲线的最低点附近,表面覆盖度几乎不随电势变化:,代入式(2),得:,(3),从微分电容曲线图可知:,(4),1 无有机物 2 达到饱和吸附 3 未达饱和吸附,-吸附量,(5),在开始吸附或脱附的电势下, d/d 变化很大,导致Cd值急剧增加,出现假电容峰。根据出现吸脱附峰的电势可粗略地估计有机物的吸脱附电势范围。,83,引起吸附过程体系自由能变化的因素,活性粒子与溶剂的相互作用。各种含极性基团的有机物(醇、酸、胺和醛等)倾向于憎水部分逸出水面,亲水部分留在溶液中,使得体系的自由能降低。,吸附层中各活性粒子间的相互作用 。可能存在各种引力(使体系自由
41、能降低)和斥力(使体系自由能升高) 。 表面活性粒子与电极表面的相互作用。包括静电作用(物理吸附)和化学作用(化学吸附)两种。,84,活性粒子与水偶极层的相互作用 。有机物吸附时,伴随着水分子的脱附,这一脱附过程使体系的自由能升高。 如果四项因素的总和使体系的自由能降低,就发生吸附(不包括离子电荷与表面剩余电荷的静电吸附)。 影响活性粒子的主要因素是:活性离子的化学性质及浓度、电极表面的化学性质和表面剩余电荷密度。因此不同活性粒子在同一电极表面,或同一活性物质在不同电极表面及不同电极电位下的吸附行为是不同的,从而表现出吸附行为的选择性。 有机分子在电极表面吸附并定向排列,取代原来在电极表面吸附
42、的水分子,形成一个新的吸附偶极子层,使电极表面剩余电荷为零时的相间电势差发生了变化。,85,有机分子吸附特点,与电极电位或电极表面剩余电荷密度有关。,在一定电极电位下,四种因素使自由能降低,发生吸附。但是随着q的增大,电容器能量迅速增加。当增加到自由能的减少不足以补偿电容器能量的增加时,有机物脱附,代之以水的吸附。所以有机分子只能在一定的电位范围内才能发生吸附。,86,零电荷电位下,电极表面剩余电荷密度为零,因而有机分子具有最大的吸附能力。吸附范围通常在零电荷电位附近。对于离子型的活性物质,还有考虑离子电荷与电极表面剩余电荷的静电作用。因而有机离子的特性吸附电位范围往往明显地向与离子电荷异号的
43、电位方向偏移。,87,与活性物质结构、浓度密切相关。饱和脂肪族化合物的表面活性在很大程度上取决于极性基团。 碳氢链数目相同的脂肪族化合物,其表面活性顺序为:羧酸胺醇脂。 同一系列脂肪族化合物的碳氢链越长表面活性越大。 对有相同碳原子数,不同支链的同素异构体来说,支链越多表面活性越小。 含有相同极性基团时,芳香族化合物的表面活性比脂肪族大,而且随苯环数的增多活性也增大很多倍。 表面活性物质的浓度增大也有利于活性粒子的吸附。,电极电位对某些有机分子吸附的排列方式也有重大影响。简单的脂肪族化合物的吸附方式没有明显变化,而芳香族和杂环化合物则在不同电位下出现两种吸附方式。,88,与电极材料有关。 电极
44、材料性质和聚集状态不同,均对活性分子的吸附有影响。同一种有机物在不同的金属电极表面的吸附行为可能有很大区别。这是由于不同金属表面的自由能不同,金属表面与活性粒子的相互作用不同及金属表面的亲水性不同所造成的。 以上讨论之涉及多半发生在“类汞电极”这种不参与电极反应的有机物吸附,对于具有催化活性的“类铂电极”,有机分子有机分子可能发生脱氢、自氢化、氧化和分解等化学反应,往往还生产一些吸附态的中间粒子。此时有机物的吸附已经不可逆。上述研究方法和规律已不再适用。,89,3 氢原子和氧原子的吸附,氢原子和氧原子吸附对金属表面电化学性质有重大影响,可以改变双电层结构和电极电位的大小,影响电极反应速度。 氢
45、原子和氧原子吸附由于在吸附的电位范围内可转变为电化学反应: 所以不再能当成理想极化电极处理 ,只能通过电量变化来研究。,90,充电曲线法,第1段氢吸附区,电极电容高达2000F/cm2,远大于双电层电容值20 F/cm2。 第2段双电层区,电极电容高达2050F/cm2。 第3段氧吸附区,电极表面形成吸附氧原子,或生成氧化物,电容值又升高了。,91,电位扫描法 氢吸附区正逆扫描均可逆; 氧吸附区不可逆。,92,氢吸附特点,低温下以分子态吸附,常温下以原子态吸附 ; 氢吸附有选择性 ; 氢吸附是可逆的; 氢吸附可分两步完成 。,93,氧吸附的特点,吸附行为很复杂, 吸附粒子泛指原子与各种含氧粒子
46、 ; 吸附过程显著不可逆性; 吸附可形成表面膜, 形成钝化效应; 氧吸附可造成与形成吸附态中间产物的电极反应速度下降。,94,3.6 双电层充电电流,3.6.1 电势阶跃,给系统突然施加一个恒定的电势E, 来看由此引起的电流 i 随时间 t 的变化。,95,在任意时间,电阻上的电压(ER)和电容上的的电压(EC)之和必定等于所施加的电压(E)。,(1),假设电容器开始未充电(在t0 时,q0),则(2)式的解:,(2),(3),对(3)微分,则得:,(4),Rs1,Cd=20F, 则20s,则在60s内,完成双层充电的95。,96,3.6.2. 电流阶跃,(1),i是常数,(5),(6),(7
47、),施加一恒定电流,97,3.6.3 电势扫描,电势从某个初始值(如E=0)开始以扫描速度(V/s)随时间线性增加,则:,(1),设t0 时,q0,(8),(9),随着电势扫描的进行,电流从零达到一个稳态值Cd,由此稳态电流可估算Cd值。,98,3.6.4 循环性电势扫描,i-t 曲线,i-E 曲线,在电势E时,扫描速度从转换到 -。,99,3.7 金属电极及电活性物质的吸附,3.7.1 金属/溶液界面层研究,液态汞电极是理想极化电极,在很大的电势窗口内是不发生氧化和还原等有电子在电极/溶液在界面间迁移的过程。而金属电极通常在表面上很容易发生氧化或还原反应(电活性物质吸附),所以难以完全满足理
48、想极化条件,一般只在非常窄的电势范围内近似的满足理想极化条件。,100,液态汞电极表面易于更新,可以保持表面洁净,金属电极表面易吸附电解质溶液中杂质而被污染,导致界面层性质的改变;,液态汞电极完全平滑,易于重现,金属电极表面在原子水平上是不平整的。,101,金属/溶液界面研究 -采用金属单晶表面作为电极,102,电容极小值对应的电势几乎不随电解质和浓度的变化而变化,表明Ag表面没有或只有弱的特性吸附。,Ag(100)晶面上测得的不同浓度KPF6和NaF的电容曲线,103,3.7.2 电活性物质的吸附,电活性物质是指那些在电极表面上能发生氧化还原反应的物质。 电活性物质的吸附能够加速电极反应,也
49、能够抑制电极反应。 氧在Pt表面吸附形成氧化膜,从而会降低电子跨越膜的电子转移速率。电活性物质的吸附在实际中有着重要的作用。 燃料电池的电催化剂,就是通过电活性物质氢气和氧气的解离吸附来加速电极反应,起到催化作用。 在Pt表面的吸附。氢和氧在Pt单晶和多晶表面的吸附研究的比较广泛和深入, 这是由于Pt是一种很好的电催化剂和研究电极,在燃料电池以及电化学研究中有着广泛的应用。,104,这是Pt单晶在0.5M H2SO4中的循环伏安图(50 mV/s),测量前首先将单晶表面在氢氧火焰中进行退火处理,退火的目的一是为了完全除去表面吸附的任何杂质,二是使单晶表面完美,即使原子完全按规则排列。 在退火处
50、理过程中,Pt表面就会有氧在表面吸附,形成氧吸附层。电势扫描时,是从开路电势OCP出发,往低电势方向扫描。,105,首先看到的这个峰是表面吸附的氧随着电势的降低而发生电还原反应所产生的还原电流所致。所以这个峰对应表面吸附氧。 第二个峰是溶液中的氢离子获得电子被还原成氢原子并吸附在Pt表面。这个峰是表面吸附的氢原子的电氧化,即这个反应的逆反应。 其它单晶上这些氧和氢的吸脱附峰同样存在,但是峰的形状和位置是不同的,这是因为表面Pt原子的排列方式是不同的。 因此,利用这些峰形的特征可以辨别晶面属于那种单晶,是100、110还是111。 通过对电流-时间曲线上的氢的吸附峰进行积分,所得的积分的面积就是
51、由于氢吸附所产成的电量(电流*时间=电量)。,106,H+ + e- Ha,QH = nF,氢吸附产生的电量,吸附氢原子的数目,表面的Pt原子的数目,每个Pt原子只吸附一个H原子,即Pt:Ha=1:1,峰形的特征可以辨别晶面类型,由对面积积分所得的电量值可以计算出吸附的氢原子的数目,显然单晶的晶面不同,QH电量不同,即吸附的氢原子的数目不同。 根据Pt单晶的晶面上原子的排列方式和原子直径的大小,可以计算出单位面积的单晶表面的Pt原子的数目。把表面Pt原子的数目和吸附的氢原子的数目比较,发现这两个值很接近.,107,电极反应速率:,由于电活性物质(H和O)的反复氧化还原吸附和脱附,表面会逐渐变得
52、粗燥,到最后单晶表面就会变成多晶表面. 多晶表面是由若干个小的不同的单晶表面组成,它反映的是这些单晶的平均行为。比如这个图就是Pt多晶(比如Pt丝,纳米Pt黑)的循环伏安图。,108,单位面积Pt多晶上吸附一层H产生的电量:,Q Pt-H = 210 C/cm2,Pt的真实表面积:,利用氢的吸脱附可测定Pt多晶电极的真实表面积 多晶是单晶的平均行为,利用H原子在Pt上的吸附和脱附研究,可以测定Pt的真实表面积。 真实电极面积对于研究电极反应动力学是一个十分重要的参数。因为我们实验能够测定的是整个电极面积上产生的电流,面积越大电流就越大,因此忽略电极面积而简单的用电流大小来表示反应速率的快慢显然是不科学和准确的。,
- 温馨提示:
1: 本站所有资源如无特殊说明,都需要本地电脑安装OFFICE2007和PDF阅读器。图纸软件为CAD,CAXA,PROE,UG,SolidWorks等.压缩文件请下载最新的WinRAR软件解压。
2: 本站的文档不包含任何第三方提供的附件图纸等,如果需要附件,请联系上传者。文件的所有权益归上传用户所有。
3.本站RAR压缩包中若带图纸,网页内容里面会有图纸预览,若没有图纸预览就没有图纸。
4. 未经权益所有人同意不得将文件中的内容挪作商业或盈利用途。
5. 装配图网仅提供信息存储空间,仅对用户上传内容的表现方式做保护处理,对用户上传分享的文档内容本身不做任何修改或编辑,并不能对任何下载内容负责。
6. 下载文件中如有侵权或不适当内容,请与我们联系,我们立即纠正。
7. 本站不保证下载资源的准确性、安全性和完整性, 同时也不承担用户因使用这些下载资源对自己和他人造成任何形式的伤害或损失。
