 病毒和进化 讲稿
病毒和进化 讲稿

《病毒和进化 讲稿》由会员分享,可在线阅读,更多相关《病毒和进化 讲稿(76页珍藏版)》请在装配图网上搜索。
1、病毒与进化病毒与进化Virus and Evolutionp 病毒的起源p 分子进化p 病毒的进化p 病毒进化与新病毒的出现p 几种重要病毒的变异与进化 假说一假说一 病毒是地球上生物进化过程中最为原始的生命 物质它产生于化学进化之后 地球生命演化的过程表现为无机物有机物化学大分子病毒原核生物真核生物;纯粹的假设假设,缺乏任何进化上的证据;假说二假说二 病毒是高级微生物的退行性生命物质 细菌类似立克茨体类的生物类似衣原体类的生物病毒;无法找到在细胞内寄生的小型细胞生物,并且在立克茨体和衣原体中未发现病毒,证据不足证据不足;“virus”一词源于拉丁文,原指一种动物来源的毒素。一词源于拉丁文,原
2、指一种动物来源的毒素。假说三假说三 病毒来源于正常细胞的核酸(内源性假说)!病毒与质粒是相似的;DNA病毒结合到细胞染色体上正好是细胞核酸外逸的逆过程;正常细胞中存在较广泛的逆转录型可动遗传因子和逆转录型重复序列,提示正常的细胞中含有RNA所介导的DNA合成反应,而这与逆转录病毒的核酸的复制行为相一致;可部分解释DNA病毒的起源,但要说明RNA病毒的起源则相当困难。1999年在古细菌古细菌中发现了所谓“反转子”的遗传单位 进化的模式如下:反转子反座子反转录转座子反转录病毒副反转录病毒DNA病毒 启示:启示:病毒起源是复杂和多元化的,不同病毒有着病毒起源是复杂和多元化的,不同病毒有着不同的起源,
3、至少在不同的起源,至少在DNA病毒和病毒和RNA病毒之间是如病毒之间是如此,这方面的结论尚有待于更多研究资料的积累。此,这方面的结论尚有待于更多研究资料的积累。达尔文的进化学说达尔文的进化学说 从生物与环境相互作用的观点出发,认为生物的变异、遗传和自然选择作用能导致生物的适应性改变。二十世纪50年代以前,对于进化的研究,主要是以古生物学、分类学、胚胎发生学、比较解剖学、生物地理学、生理学和群体遗传学等方面为主。分子进化分子进化 分子生物学和分子遗传学的兴起使得进化学家开始从分子水平来研究生物进化的原理和机制。在核酸和蛋白质分子组成的序列中,蕴藏着大量生物进化的遗传信息。生物大分子的进化 基因或
4、生物个体的演化历史的重构 分子进化研究有助于进一步阐明物种进化的分子基础,探索基因起源机制,从基因进化的角度研究基因序列与功能的关系。系统发育分析可用于示踪快速变化的物种(如病毒、细菌系统发育分析可用于示踪快速变化的物种(如病毒、细菌等),对于种群内变化类型的分析可以说明一个特定基因是否处等),对于种群内变化类型的分析可以说明一个特定基因是否处于选择压力之下,这是流行病学应用的重要信息。于选择压力之下,这是流行病学应用的重要信息。事实事实1 1:生物都有过度繁殖的倾向:生物都有过度繁殖的倾向事实事实2 2:物种内的个体数能保持稳定:物种内的个体数能保持稳定事实事实3 3:资源是有限的:资源是有
5、限的推论推论1 1:个体间存在着生存斗争:个体间存在着生存斗争推论推论1 1:个体间存在着生存斗争:个体间存在着生存斗争事实事实4 4:个体间普遍存在差异(变异):个体间普遍存在差异(变异)事实事实5 5:许多变异是可以遗传的:许多变异是可以遗传的推论推论2 2:具有有利变异的个体,生存并:具有有利变异的个体,生存并留下后代的机会多留下后代的机会多推论推论3 3:有利变异逐代积累,生物不断进化出新类型:有利变异逐代积累,生物不断进化出新类型过度繁殖过度繁殖生存斗争生存斗争遗传变异遗传变异适者生存适者生存 达尔文的自然选择学说达尔文的自然选择学说 综合进化论(现代达尔文主义)综合进化论(现代达尔
6、文主义)种群种群是生物进化的单位,进化机制研究属于群体遗传学范畴。突变和基因重组突变和基因重组产生进化的原材料。自然选择自然选择决定生物进化的方向。隔离隔离导致物种的形成。引起种群基因频率改变的因素引起种群基因频率改变的因素A.突变突变 方向是随机的,给自然选择提供了原材料。如果突变性状被选择,这一突变基因就在基因库中积累增多。B.遗传漂变遗传漂变 在一个小种群内,基因频率由于偶然的机会(不是自然选择的原因)而随机增减的现象。一个种群中的几个或几十个个体迁移到另一地区而定居下来,自行繁衍后代,造成基因频率发生改变的现象。C.基因迁移基因迁移 一个种群的个体迁入到另一个种群中去的现象。不管是老个
7、体的迁出还是新个体的加入,都会使种群基因频率发生变化。D.自然选择自然选择 通过自然选择对基因型的影响和基因重组的作用,从而定向改变群体的基因频率。结果使生物类型发生改变。青霉素对细菌抗性的选择 DDT对家蝇抗性的选择E.选择压选择压 指在2个相对性状之间,其中一个性状被选择而生存下来的优势。它是经选择以后发生效果的一个标准。通过比较不同生物的某些功能相同的蛋白质或核酸的氨基酸或核通过比较不同生物的某些功能相同的蛋白质或核酸的氨基酸或核苷酸序列的差异,与物种的表型进化情况基本一致。苷酸序列的差异,与物种的表型进化情况基本一致。分子进化至少有分子进化至少有3个显而易见的特点:个显而易见的特点:一
8、是多样性程度高,与表型多态一是多样性程度高,与表型多态(即在一相互交配的群体中存在着两种(即在一相互交配的群体中存在着两种或多种基因型的现象)或多种基因型的现象)相比,分子多态更为丰富相比,分子多态更为丰富(例如细胞色素(例如细胞色素C在有氧呼在有氧呼吸的不同物种中就有种种不同的分子结构;乳酸脱氢酶吸的不同物种中就有种种不同的分子结构;乳酸脱氢酶LDH可形成可形成5种种LDH同同工酶:工酶:H4、M4、H3M1、H2M2和和HM3,其中,其中M型在骨骼肌,型在骨骼肌,H型在心肌)型在心肌);二是各种二是各种同源同源分子对选择大都是中性或近中性的,它们都有完整的分子对选择大都是中性或近中性的,它
9、们都有完整的高级结构,能很好地完成各自的功能高级结构,能很好地完成各自的功能(如脊椎动物的血红蛋白分子都能运(如脊椎动物的血红蛋白分子都能运氧、各种生物的细胞色素氧、各种生物的细胞色素C都能在氧化磷酸化中完成电子的传递等)都能在氧化磷酸化中完成电子的传递等);三是随着生物从低级向高级演化,三是随着生物从低级向高级演化,同源同源分子中逐年发生氨基酸或核分子中逐年发生氨基酸或核苷酸的替换,且大致按每年每位置替换数恒定速率进化苷酸的替换,且大致按每年每位置替换数恒定速率进化;由此说明,自然群体在分子水平上存在着意想不到的多态性,而由此说明,自然群体在分子水平上存在着意想不到的多态性,而这正是综合进化
10、论所解释不通的。这正是综合进化论所解释不通的。1968年,日本的木村资生提出了分子进化的中性学说年,日本的木村资生提出了分子进化的中性学说 分子钟分子钟 某一蛋白在不同物种间的取代数与所研究物种间的分歧时间接近正线性关系,进而将分子水平的这种恒速变异称为“分子钟”。The protein,haemoglobinFishGoosePigHumanWormLooking at beta-globinThe structure of a human haemoglobin moleculeA human beta-globin(Hbb)unitAll of the animals pictured
11、above produce beta globin.It follows that they all must have a gene for beta globin.We can look at relationships between these animals by comparing their beta globin genes.分子的异速进化现象被广泛的观察到分子的异速进化现象被广泛的观察到 例如:例如:分子序列证据与化石证据在人类起源时间上的差异分子序列证据与化石证据在人类起源时间上的差异 分歧3000万年的物种氨基酸序列差异的应达45%、非重复序列DNA差异应约为8%,但实测
12、值分别为0.8%与1.1%,存在6倍左右的差别。许多人类学家倾向于怀疑钟的存在,并认为在高等灵长类中分子进化速率下降。总之,虽然大部分分子进化学家同意序列进化与分歧时间密相关,但进化是以年限年限还是以代限代限为刻度则仍有分歧与争议;而且因为众多因素的影响,与分子钟相左的数据,无论是用氨基酸、核苷酸序列差异、免疫学距离,还是用DNA杂交复性等参数,均不断有所报道,其论争预计将继续下去。分子进化中性学说分子进化中性学说分子进化速度的一定性,即同一生物大分子在不同物种中的进化速度是一样的;功能上对生命生存制约性低的分子或一个分子中不那么重要的部分,较之对生命生存制约性高的分子或分子中重要的部分,其突
13、变置换率高;进化过程中,对分子功能不损害或损害轻的突变(置换)较之损害严重的突变容易发生;具有新功能的基因一般起源于基因重复;中性突变包括有害程度轻微的突变;分子进化中遗传漂变对中性突变在群体中的固定发挥着重要作用,即遗传漂变是分子进化的基本动力;无法解释物种的形成;无法解释物种的形成;无法解释表现型水平上的进化;无法解释表现型水平上的进化;中性学说面临的中性学说面临的两大难题两大难题比较现代达尔文主义和中性学说对生物进化解释的差异。生物进化的主导因素生物进化的方向影响生物进化的速率因素现代达尔文主义中性学说自然选择中性突变本身随机自由组合环境和生物世代恒定的自然选择 达尔文主义和中性学说是两
14、种完全不同的机达尔文主义和中性学说是两种完全不同的机制,如何实现这两者的统一:制,如何实现这两者的统一:分子水平上的进化主要是由中性突变与遗传漂变导致的分子水平上的进化主要是由中性突变与遗传漂变导致的基因频率的固定所造成的;表现型水平上的进化则是适应基因频率的固定所造成的;表现型水平上的进化则是适应性进化,自然选择对此是最合理的解释。性进化,自然选择对此是最合理的解释。“中性进化恐怕不应该说是非达尔文式进化,而是应该中性进化恐怕不应该说是非达尔文式进化,而是应该说是进化中间的非达尔文式变化。说是进化中间的非达尔文式变化。”两者关键在于对选择对象的解释不同。两者关键在于对选择对象的解释不同。中性
15、学说中性学说:基因即碱基对,分子进化:基因即碱基对,分子进化 达尔文主义达尔文主义:个体,种群进化:个体,种群进化在理解生物进化上可把中性学说看作是附加在自然选择学在理解生物进化上可把中性学说看作是附加在自然选择学说中的一个原理,是在分子水平上对达尔文主义的补充和说中的一个原理,是在分子水平上对达尔文主义的补充和发展。发展。序列对位排列序列对位排列 在序列对位排列时,通过插入间隔(在序列对位排列时,通过插入间隔(gap)的的方法使不同长度的序列对齐,达到长度一致。优方法使不同长度的序列对齐,达到长度一致。优化的对位排列应使间隔的数目最小,同时序列间化的对位排列应使间隔的数目最小,同时序列间相似
16、区域最大。相似区域最大。X:CGATCAGY:CC-TCAGX:CG-ATCAGY:CG-TCAGZ:CGGAATCAG 起始空位和延伸空位罚分(Gap Openning and Gap extension Penalties)在序列比对过程中无限地引入空位可使两个序列具有最高的相似性,但却无生物学意义,因此引入空位罚分。对于起始空位的罚分与延伸空位不同。X:-AGCTGAG-G-AC-G-AC Y:CAGCTGA-CCTGCACAGTAC 多数比较涉及全长序列的比较,因此不能简单理解为如何多数比较涉及全长序列的比较,因此不能简单理解为如何减少间隔的数目,还要考虑对位排列后的生物学意义。减少间
17、隔的数目,还要考虑对位排列后的生物学意义。记分矩阵(记分矩阵(scoring matrix)scoring matrix)广泛用于评价序列对位排列广泛用于评价序列对位排列的质量。通常用得分(的质量。通常用得分(+)、无分()、无分(0 0)或罚分来综合评价。)或罚分来综合评价。取代矩阵取代矩阵 上述比对方法仅仅用相同或不同来描述两个上述比对方法仅仅用相同或不同来描述两个残基的关系,而取代矩阵考虑到特定位置不同性质的氨基残基的关系,而取代矩阵考虑到特定位置不同性质的氨基酸取代对蛋白质的结构与功能会产生截然不同的影响。如酸取代对蛋白质的结构与功能会产生截然不同的影响。如异亮氨酸(异亮氨酸(Isol
18、eucineIsoleucine)和缬氨酸()和缬氨酸(VaineVaine)(体积小,疏体积小,疏水水)、丝氨基酸和苏氨酸(极性)之间可以很能容易地相、丝氨基酸和苏氨酸(极性)之间可以很能容易地相互取代而不用改变它们的生理生化性质,而谷氨酸对异亮互取代而不用改变它们的生理生化性质,而谷氨酸对异亮氨酸的取代则会对蛋白的理化性质产生重要影响,对这两氨酸的取代则会对蛋白的理化性质产生重要影响,对这两类不同取代应给予不同的打分,可大大提高比对的敏感性类不同取代应给予不同的打分,可大大提高比对的敏感性和生物学意义。和生物学意义。Dayhoff突变数据矩阵:点突变记分矩阵称为MD(mutation da
19、ta)或PAM(point accepted mutation)矩阵。一个PAM进化距离为每100个氨基酸中一个点突变被接受的概率。适用于相似性较高的序列比对(通常达85%以上),其后的数字表示进化距离的大小,数值越小,相似性越高。而进化距离较远的矩阵如PAM250的准确性受到限制。大于大于0 0的值表示突变的可能性大,等于的值表示突变的可能性大,等于0 0是中性的是中性的(随机突变),小于(随机突变),小于0 0则表示突变的可能性较小则表示突变的可能性较小。BLOSUM(BLOCKS Substitution Matrix),用于远源序列间的分析。高于或等于80%相同残基的序列组成的模块组可
20、用BLOSUM80矩阵来构建,BLOSUM62不同记分方法的特点不同记分方法的特点 1.基于一致性的记分基于一致性的记分(高度相似,不一致无体现)高度相似,不一致无体现)2.基于化学相似性的记分基于化学相似性的记分 3.基于遗传密码的记分。基于遗传密码的记分。4.基于观察的记分。基于观察的记分。点标方法点标方法 点标法是两序列比较最基本和最直观的方法 全局排列和局部排列全局排列和局部排列 序列对位排列可分为全局序列对位排列可分为全局(global alignment和局和局部排列部排列(local aglinment)两大类两大类 全局排列是对序列的全长进行最优对位排列全局排列是对序列的全长进
21、行最优对位排列,由由Needleman和和Wunsch于于1970年提出年提出.局部对位排列由局部对位排列由Smith和和Waterman于于1981年提年提出出,以在局部区域内寻找最优的对位排列以在局部区域内寻找最优的对位排列.序列对位排列的用途序列对位排列的用途:1.构建系统发育树构建系统发育树;2.结构预结构预测测;3.序列基序鉴定序列基序鉴定;4.功能预测功能预测;5.数据库搜索数据库搜索;6.起源或进化分析起源或进化分析;系统发生树系统发生树(phylogenetic tree)是描述一群有机体发生或是描述一群有机体发生或进化顺序的拓扑结构进化顺序的拓扑结构 进化树给出分支层次或拓扑
22、图形,它是产生新的基因复制或享有共同祖先的生物体的歧异点的一种反映,树枝的长度反映当这些事件发生时就存在的蛋白质与现在的蛋白质之间的进化距离。有权值的树(或标度树,scaled tree,树中标明分支的长度);无权值树(或非标度树,unscaled tree);LeafLeafLeafLeafLeafBranchBranchNodeRootBranchROOTED PHYLOGENETIC TREELeafLeafLeafBranchUNROOTED PHYLOGENETIC TREE系统发育树重建分析步骤系统发育树重建分析步骤12345Sequence dataAlign Sequences
23、Phylogenetic signal?Patternsevolutionary processes?Test phylogenetic reliabilityDistances methodsChoose a methodMBMLCharacters based methodsSingle treeOptimality criterionCalculate or estimate best fit treeLSMENJDistance calculation(which model?)Model?MPWheighting?(sites,changes)?Model?Alignment-bui
24、lding the data model and extracting a dataset多序列比对(自动、人工)多序列比对(自动、人工)Determining the substitution model-consider sequence variation 建立取代模型(建树方法)建立取代模型(建树方法)Tree building建立进化树建立进化树Tree evaluation进化树评估进化树评估系统发育树重建的基本方法系统发育树重建的基本方法系统发生树的构建方法分为两大类:基于距离距离的构建方法 非加权组平均法(Unweighted Pair Group Method with Ar
25、ithmetic mean,UPGMA)邻近归并法(Neighbor Joining)Fitch-Margoliash法 最小进化方法 基于离散特征离散特征的构建方法 最大简约法 最大似然法 进化简约法 相容性方法 基于距离的系统发生树构建方法基于距离的系统发生树构建方法 给定一种序列之间距离的测度,在该距离测度下构建一棵系统发生树,使得该树能够最好地反映已知序列之间的距离。计算序列的距离,建立距离矩阵通过距离矩阵建进化树10条核酸序列的距离矩阵 根据距离矩阵构造系统发生树根据距离矩阵构造系统发生树 连锁聚类方法及非加权分组平均法连锁聚类方法及非加权分组平均法 选择距离最小的一对序列 将这两个
26、序列合二为一,形成一个新的对象(代表这两个序列的祖先)重新计算这个新的对象与其它序列的距离。假定的前提条件是:在进化过程中,核苷酸或氨基酸的替换速率是均等且恒定的,在每一次分歧发生后,从共同祖节点到两个分类单元间的分支长度一样。在构建系统发生树时,首先用n个叶节点表示n个分类单元(序列),每个分类单元自成一类,然后通过反复的聚类使所有的分类单元都聚为一类,并将进化过程中的祖先赋予树的内部节点,最终得到一个完整的系统发生树。得到一棵有根的系统发生树,从树根到任何叶节点的分支长度全都一样,即所有物种的突变速率相同,存在一个固定节律的“分子钟分子钟”,各个物种从树根出发,踏着同样的节律,沿不同的路径
27、,演化成为当前的形式。非加权分组平均法非加权分组平均法 在平均连锁聚类过程中,一个新类到其它类之间的距离就是简单的原距离平均值。这样的计算非常简单,但是,如果各个类中分类单元个数不一样,原距离矩阵中各个距离值对新距离计算的贡献就不一样,或者说是经过“加权”的,称这样的聚类为加权分组平均(Weighted pair group method with Arithmetic mean,WPGMA)。在非加权分组平均法中,在计算新分类到其它分类之间的平均距离时按照各分类中分类单元的数目进行加权处理。UPGMA算法的执行过程如下:初始化:使每个物种自成一类,如果有n个物种,则开始时共有n个类,每个类的
28、大小为1,分别用n个叶节点代表每个类;执行下列循环:I.寻找具有最小距离Dij的两个类i、j;建立一个新的聚类(ij)II.连接i和j形成新节点(ij),生长两个新的分支,将i 和j 连接到(ij),分支的长度为Dij/2;III.计算新分类到其它类的距离 其中ni、nj、(ni+nj)分别为i类、j类、(ij)类的元素个数;l 在距离矩阵中删除与类i和类j相应的行和列,为类(ij)加入新的行和列;重复循环,直到仅剩一个类为止。重复循环,直到仅剩一个类为止。kjjijkijiikijDnnnDnnnD,),()()(d=e=10/2=5c=19/2=9.5g=c-d=9.5-5=4.5AB(C
29、DE)A-2239.5B-41.5(CDE)-a=b=22/2=11(AB)(CDE)(AB)-40.5(CDE)-f1+a=f2+c=40.5/2=20.25f1=9.25,f2=11.75 Fitch-Margoliash方法方法 找出关系最近的序列对,如D和E 将剩余的序列作为一个简单复合序列,分别计算D、E到所有其他序列的距离的平均值 用这些值来计算D和E间的距离 将D、E作为一个单一的复合序列DE,计算与每一个其他序列的距离,生成新的距离矩阵 确定下一对关系最近的序列,重复前面的步聚计算枝长 从每个序列对开始,重复整个过程 对每个树计算每对序列间的预测距离,发现与原始数据最符合的树D
30、E距离=d+e D到ABC间的平均距离=d+mE到ABC间的平均距离=e+m d=4,e=6c+g+(e+d)/2=19 c+f+(a+b)/2=40(e+d)/2+(a+b)/2+f+g=41得:c=9 基于特征的系统发生树构建方法基于特征的系统发生树构建方法分类单元特征矩阵分类单元分类单元 位点位点1 1 位点位点2 2 位点位点3 3 位点位点4 4 位点位点5 5 位点位点6 6 甲甲 C A G G T A 乙乙 C A G A C A 丙丙 C G G G T A 丁丁 T G C A C T 戊戊 T G C G T A 一般问题:给定n个物种 m个用以描述物种的特征 每个物种所
31、对应的特征值 构建一棵系统发生树,使得某个目标函数最大。最大简约法 最大似然法 进化简约法 相容性方法 系统发生树的可靠性系统发生树的可靠性 在距离法中,连锁聚类方法比较简单,非加权分组平均法比较实用,当使用的距离数据是来源于多个基因的分析结果时,利用非加权分组平均法能得到可靠的系统发生树。对于离散特征分析方法,如果序列趋异程度较小、核苷酸替换的速率或多或少的恒定,最大简约法是一种较好的系统发生树构建法。无论是基于距离的系统发生树重建方法,还是基于特征的系统发生树重建方法,都不能保证不能保证一定能够得到一棵描述比对序列进化历史的真实的树。总规则是总规则是 用截然不同的距离矩阵法和简约法分析一个
32、数据集,如果能够产生相似的系统树,这样的树可以被认为是相当可靠的。我们需要评价一棵系统发生树的可靠性,这涉及两个问题,即整棵树我们需要评价一棵系统发生树的可靠性,这涉及两个问题,即整棵树和它的组成部分(分支)的置信度是多少?和它的组成部分(分支)的置信度是多少?自举检验自举检验(bootstrap test)基本方法是:基本方法是:从原数据集中抽取(同时替换)部分数据组成新的数据集,然后用这个新的数据集构造系统发生树。重复该过程,产生成百上千的重采样数据集,并同时生成对应的自举树,进而检验自举树对最终系统发生树各个分支的支持率。具体做法是,将最终系统发生树与各个自举树进行比较,其中,在各个自举
33、树中都有出现或大量出现的那些部分将具有较高的置信度。产生相同分组的自举树的数目常常标注在系统发生树相应节点的旁边,表示树中每个部分的相对置信度。目前已经发现许多蛋白的一级序列差异很大,难以通过序列比对进行分子进化的研究,但它们的空间拓扑结构仍然很相似,可以进行结构叠合比较、分析它们之间的进化关系,这表明结构比较可以比序列比较获得更多更精确的结构信息。结构进化树结构进化树 刚体结构叠合比较刚体结构叠合比较 用比较后确定的拓扑等价位点的个数或等价位点C原子距离的均方根值作为不同结构间差异性的量度;多特征的结构比较多特征的结构比较 用蛋白质结构的多项特征如残基的物理特性、残基的空间倾向性、主侧链的方
34、向、主链的二面角、二级结构类型和主侧链的可接近性等综合指标作为结构的差异性量度,有时称此类方法构建的结构进化树为“类结构”进化树。不同构树方法的特点方法方法优点优点缺点缺点距离法距离法 速度快,稳健,构建唯一系统发育树序列转化为距离时信息量有丢失。对分歧明显的序列,很难对距离进行可靠估计简约法简约法 如果树枝短,序列相似性高,信息位点多,结果可靠树枝长度变异大时可靠性较低似然法似然法 以似然率反映数据最支持怎样的系统发育关系搜索所有可能的系统发育树计算量大 系统发生分析常用软件系统发生分析常用软件http:/evolution.genetics.washington.edu/phylip/so
35、ftware.html 病毒进化的机制病毒进化的机制 基因突变是其进化和突变的基因突变是其进化和突变的基础基础 病毒的复制每日数以亿计 RNA病毒转录酶不具有校对功能 DNA病毒聚合酶有纠错功能,其突变率远低于RNA病毒,但也大大超过宿主细胞 如果该处核苷酸分别参与重叠的开放读码框,则单一位点的突变可改变2个开放读码框 病毒在不同的组织中突变速率不同 病毒基因组中不同部分的突变速率不同 基因交换基因交换加快加快病毒的进化病毒的进化 高速的复制及高度的突变率增加了病毒的基因多样性,高速的复制及高度的突变率增加了病毒的基因多样性,为其突变和进化提供了首要条件。为其突变和进化提供了首要条件。病毒之间
36、可以通过基因组片段的重组或重配交换遗病毒之间可以通过基因组片段的重组或重配交换遗传信息,这是病毒在自然选择过程中继续生存所必需。传信息,这是病毒在自然选择过程中继续生存所必需。快快不管病毒是怎样起源的,它在生物学上是极其成功的,不管病毒是怎样起源的,它在生物学上是极其成功的,因为没有生物可以逃逸它的侵染。因为没有生物可以逃逸它的侵染。病毒进化的两条途径病毒进化的两条途径 病毒与其宿主共同进化共同进化,二者具有共同的命运。这样,病毒将面临生存的瓶颈,即病毒可能因为宿主的消亡或因抗病毒措施的应用而消亡;一般而言,DNA病毒病毒进化依照第一条途径,病毒选择多种宿主多种宿主,从而具有较广阔的生存环境。
37、当一种宿主面临危险时,病毒可以在另一种宿主中繁殖。大多数RNA 病毒病毒的进化通过第二种途径。这两种途径都为病毒提供了最佳的生存方式。病毒进化的方向病毒进化的方向 突变是随机发生的,而病毒进化则可能有方向性突变是随机发生的,而病毒进化则可能有方向性 负性选择负性选择 适应性选择适应性选择 免疫选择免疫选择 药物选择药物选择 RNA作为遗传物质在病毒进化中的优越性作为遗传物质在病毒进化中的优越性 可能可能由于由于RNARNA在信息传递中具有较高的可塑性而可使病毒能更在信息传递中具有较高的可塑性而可使病毒能更好地适应各种不同环境,故多数病毒用好地适应各种不同环境,故多数病毒用RNARNA作为自己的
38、遗传物质。作为自己的遗传物质。病毒突变与进化方式病毒突变与进化方式 单寄主一般突变与进化 变寄主基因突变与进化 所谓的新病毒,除了少数是在本身寄主中突变产生的,其余大所谓的新病毒,除了少数是在本身寄主中突变产生的,其余大部分都是因为寄主的改变而产生的。部分都是因为寄主的改变而产生的。污染促进突变理论污染促进突变理论 自然环境的化学污染在不断加速,生态系统中污染物的浓度和种类在日益增加,对病毒这样的简单生物来说,多个污染物的联合效应可导致其加速进化,并在一、二十年(技术时间水平)内完成这种大幅度的进化。抗生素、合成农药和激素等抗生素、合成农药和激素等 SARSSARS起源于污染对病毒进化的加速诱
39、导?起源于污染对病毒进化的加速诱导?生物性复合污染生态效应生物性复合污染生态效应 抗病毒治疗对病毒突变的影响抗病毒治疗对病毒突变的影响 药物改变了宿主体内环境,打破了平衡,诱导了病毒的突变药物改变了宿主体内环境,打破了平衡,诱导了病毒的突变方向,形成药物选择。方向,形成药物选择。病毒突变带来的新问题病毒突变带来的新问题 耐药株的出现耐药株的出现 检测试剂的失效检测试剂的失效 传统疫苗的失败传统疫苗的失败 新的病毒性疾病的出现新的病毒性疾病的出现Hepatitis C 新现病毒的特点新现病毒的特点 几乎所有新现病毒的基因组均为RNA而非DNA,出现这种情况的原因在于这两种类型致病因子的进化速率进
40、化速率不同;几乎所有的新现病毒都有一个动物储存库动物储存库,其出现的过程往往被归因于跨种属跨种属传播;原因原因 供体和受体种群的亲近度亲近度改变,从而增加了人类与动物病原接触的机会;受体和供体的数量和密度数量和密度改变,增加的病毒一旦进入新的物种,就有可能形成持续传递的网络;病毒的变异变异;冠状病毒进化关系分析冠状病毒进化关系分析目的目的 分析冠状病毒内部的亲缘关系背景背景 一种新型的冠状病毒(SARS-CoV)被认为是SARS的病原体;与人和动物的许多疾病疾病有关;根据其基因组和抗原性特征,已知冠状病毒可大致分为三组三组:group I 来自猪等动物,如猪传染性胃肠炎病毒、人冠状病毒229E
41、;group 来自牛、鼠等动物,如牛冠状病毒、小鼠肝炎病毒、人冠状病毒OC43;group 来自鸡等禽类,如火鸡蓝冠病毒、鸟类传染性支气管病毒;人类的冠状病毒种类比较多,分别属于第一组和第二组。引起普通感冒的病毒中有20%属于冠状病毒,但普通感冒症状较轻,因此不像SARS冠状病毒引人注目。方法方法 原始数据原始数据 24个冠状病毒全基因组序列从个冠状病毒全基因组序列从GeneBank下载下载,进化距离算法进化距离算法 构建进化树构建进化树 使用使用PHYLIP软件包中的软件包中的UPGMA方法对该距离方法对该距离进行聚类,最终的构建进化树进行聚类,最终的构建进化树结果结果 SARS 冠状病毒属
42、于一个冠状病毒属于一个新新组;组;SARS 冠状病毒经历了一个冠状病毒经历了一个独立独立的进化路径;的进化路径;SARS 冠状病毒的前体可能存在冠状病毒的前体可能存在于某一于某一特定的宿主特定的宿主内,并且内,并且独独立进化立进化了很长时间了很长时间;Evolutionary Insights into the Ecology of Coronaviruses目的目的 The identification of closely related viruses in animal hosts is a prerequisite for establishing the ecology of vi
43、ral emergence along with reconstruction of evolutionary pathways;however,complex ecosystems make this difficult.This information is critical for the control of these diseases at their sources and for intervention in possible outbreaks.方法方法Sampling and virus detection.Shenzhen and Guangzhou,China.The
44、 helicase(HEL)domain of the replicase gene,the spike protein(S)gene,and the nucleocapsid protein(N)gene were chosen for analysis.Phylogenetic analysis.Sequence alignments using TransAlign with ClustalX Phylogenetic trees using neighbor joining in PAUP*.Detection of recombination and lineage-specific
45、 selection.using a genetic algorithm for recombination detection(GARD)Estimation of divergence dates and population dynamics.using the uncorrelated relaxed clock model in BEAST.Commom cold结果结果 Phylogenetic relationships of coronaviruses.HEL the most suitable for calculation;SARS-CoVs and SARS-like B
46、tCoVs;common cold coronaviruses and PEDV;wild animal coronaviruses(raccoon dog CoV/GZ43/03 and Chinese ferret badger CoV/DM95/03)along with canine and feline coronaviruses and TGEV;BtCoVs are precursors to previously established coronavirus lineages.nucleocapsid helicasespike Estimation of divergenc
47、e dates.Based on HEL domain sequence.Breda virus)was used to root the tree.Detection of recombination and lineage-specific selection.Five recombination breakpoints were detected;no interhost recombination was observed in the phylogenies;These findings further support the hypothesis that SARS-CoVs we
48、re recently introduced into civet and human populations from an unknown host.Population dynamics of coronaviruses.Analysis of population dynamics revealed that coronavirus populations in bats haveconstant population growth,while viruses from all other hosts show epidemic-like increases in population
49、.Taken together,these results indicate that coronaviruses are endemic in bat populations and that after interspecies transmission to a naive host,there was a change to epidemic-like population growth,particularly for group 4B,which contains civet and human SARS-CoVs.Bats are the natural reservoir of
50、 all currently known coronavirus lineages.Bats are likely the natural hosts for all presently known coronavirus lineages and that all coronaviruses recognized in other species were derived from viruses residing in bats.Diverse coronaviruses are endemic in different bat species,with repeated introduc
51、tions to other animals and occasional establishment in other species.The genomic and epidemiological dynamics of human influenza A virusComplete genome sequences of A/H3N2 and A/H1N1 influenza viruses fromNew York state,USA,and New Zealand,were collated from the NCBI Influenza Database as part of th
52、e Influenza Genome Sequencing Project Avian Influenza Virus Exhibits Rapid Evolutionary Dynamics背景背景Natural reservoirs of influenza A viruses are wild aquatic birds(with the exception of H5N1 highly pathogenic avian influenza virus);,A key concept in influenza virus evolution is that thereis a marke
53、d difference in evolutionary dynamics betweenthose viruses that infect aquatic birds and those from otherhost species.The substitution rates estimated for AIV were very high,ranging from 1.8 to 8.4103 substitutions per site,per year(subs/site/year).previously estimated in human(5.7103 subs/site/year
54、 in the HA1 domain;equine(5.4103 and 5.1103 subs/site/year for the M and NS genes,respectively;swine(1.30103 subs/site/year for the M gene Therefore indicate that AIV does not evolve anomalously slowlyAge of the MRCAs of avian influenzaA virusesThe age of the MRCA of each gene and serotype was gener
55、ally extremely recent(range 15.4196.0 years,with most less then 100 years).Segments taken from H5N1 viruses often have very shallow genetic diversity(21.0101.9 years).These dates are still substantially older than the first appearance of H5N1 in humans in 1997,and although this subtype was first iso
56、lated in 1959,the majority of isolates analyzed were sampled after 1996.To determine the factors that might affect the pattern of AIV evolutionno significant difference in substitutionrates among genes.Similarly,there was little differencein evolutionary rates among serotypes,The highest dN/dS ratio
57、s were observedin the HA and NA(mean dN/dS 0.130 and 0.206,respectively),most likely reflecting immune selection pressureat a small number of amino acid sites(Horimoto andKawaoka 2005),and also NS1(mean dN/dS 0.147),whichdownregulates dsRNA-induced antiviral responses.H3H1B phylogenies constructed u
58、sing|the HA1 domain However,providing a biological explanation for these differences is complicated;The diversity of a pathogen population is determined by the balance between the production of new strains and the competition-induced stochastic extinction of existing variants;To explainTo explain th
59、e surprisingly limited genetic diversity of influenza,Explore four determinants of this balance:the of new variants through mutation or reassortment the initial of those variants in the host population-mediated competition between strains the processes governing the rate of strain Unifying the spati
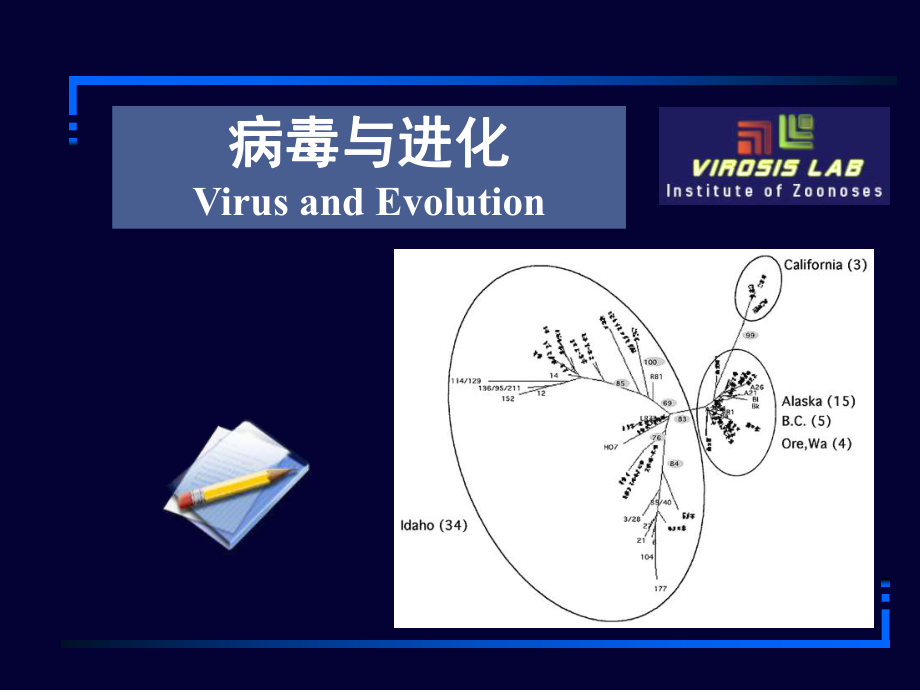
60、al population dynamics and molecular evolution of epidemic rabies virus目的目的 Linking ecological dynamics of infectious disease with underlying molecular and evolutionary change.背景背景 The past two decades have witnessed an expanding list of infectious diseases characterized as emerging threats to human
61、 andor wildlife health,agricultural production,or public security;Infectious disease emergence is under the simultaneous influence of both genetic and ecological factors;For example,SARScoronavirus can be understood as a combined ecological and evolutionary phenomenon,the appearance of a novel genet
62、ic type of coronavirus coupled to the movement and dispersal of infected individuals over transportation networks;方法方法 Rabies Phylogeny and Genetic Distances.83 individual fox rabies samples collected in Ontario Sequences were aligned by using the CLUSTALW Maximum likelihood(ML)phylogenetic analysis sequences A ML tree was found in a heuristic search in PAUP under a selected model Spatial Pattern Analysis.Correlations between pair-wise genetic and pair-wise geographic distances were performed by using standard regression techniques
- 温馨提示:
1: 本站所有资源如无特殊说明,都需要本地电脑安装OFFICE2007和PDF阅读器。图纸软件为CAD,CAXA,PROE,UG,SolidWorks等.压缩文件请下载最新的WinRAR软件解压。
2: 本站的文档不包含任何第三方提供的附件图纸等,如果需要附件,请联系上传者。文件的所有权益归上传用户所有。
3.本站RAR压缩包中若带图纸,网页内容里面会有图纸预览,若没有图纸预览就没有图纸。
4. 未经权益所有人同意不得将文件中的内容挪作商业或盈利用途。
5. 装配图网仅提供信息存储空间,仅对用户上传内容的表现方式做保护处理,对用户上传分享的文档内容本身不做任何修改或编辑,并不能对任何下载内容负责。
6. 下载文件中如有侵权或不适当内容,请与我们联系,我们立即纠正。
7. 本站不保证下载资源的准确性、安全性和完整性, 同时也不承担用户因使用这些下载资源对自己和他人造成任何形式的伤害或损失。
